
Inhibition of complement dependent cytotoxicity by anti-CD20 aptamers†‡
Nadia L. Al-Youssef,
Shahrokh M. Ghobadloo and
Maxim V. Berezovski *
*
Department of Chemistry and Biomolecular Sciences, University of Ottawa, 10 Marie Curie, Ottawa, K1N 6N5, Canada. Web: http://mysite.science.uottawa.ca/mberezov/E-mail: maxim.berezovski@uottawa.ca
First published on 26th January 2016
Abstract
Complement dependent cytotoxicity (CDC) plays a vital role in human immunity and is important for the action of therapeutic antibodies. In addition, the deregulation of complement dependent cytotoxicity is a main contributor to autoimmune diseases and inflammatory pathologies, including, rheumatoid arthritis, lupus and transplant rejection. In this work, we developed DNA aptamers inhibiting complement dependent cytotoxicity in vitro. Aptamer clones selected by differential cell-SELEX and next-generation sequencing technologies possessed strong affinity (Kd < 75 nM) and selectively to B-lymphocyte antigen CD20 and showed good results in competitive displacement of CD20 antibodies. More importantly, the anti-CD20 aptamers protected and increased the viability of cells in the presence of CD20 antibodies and complement factors from human serum. These shielding aptamers can serve as drug candidates for inhibition of complement activation in the treatment of autoimmune diseases and organ transplantation.
CD20 is a cell-surface antigen expressed on pre-B and mature B-lymphocytes as well as on a variety of B-cell lymphomas. It is a component of a signal transduction complex involved in the growth and differentiation of B-cells.1 CD20 is the target of several monoclonal antibodies (mAbs), like rituximab, ofatumumab, obinutuzumab, ibritumomab tiuxetan, and tositumomab, used in the clinic in the treatment of B-cell malignancies.2 Anti-CD20 antibodies have various effector functions, among them the promotion of Complement Dependent Cytotoxicity (CDC) in target cells.3 The complement system is a major activator and facilitator of innate and acquired immunity.4 It is responsible for the opsonization and elimination of pathogenic organisms, as well as the clearance of immune complexes and apoptotic cells.5 CDC relies on the cascade-like activation of more than 30 blood-borne and cell-based factors, culminating in the formation of a membrane attack complex (MAC).6 The MAC triggers numerous intracellular signals,7 or inflict damage leading to necrotic cell death.8 Under a variety of conditions, complement activation leads to rapid and highly effective destruction of either foreign pathogens or normal tissue, causing autoimmune diseases or organ rejection after transplantation. Therefore, to find molecular agents inhibiting CDC is as important as to find molecules activating CDC.
In this work, we selected DNA aptamers inhibiting complement dependent cytotoxicity. The synthetic aptamers acted as antagonists of anti-CD20 mAb, and suppressed CD20 mAb-mediated CDC. Before starting the selection we obtained CD20 positive and negative cells by expressing CD20 in a HEK293T cell line using a commercially available lentiviral transfection system. The CD20 expression was evaluated using flow cytometry and fluorescent microscopy (Fig. S1 and S2†). The positively transfected cells, CD20+HEK, and the wild-type CD20 negative HEK cells were used in a differential cell-SELEX protocol.9
In order to isolate CD20 specific aptamers, we began by incubating a fluorescently labelled “Harvard” DNA library with the CD20+HEK cells (Fig. 1). It is called the Harvard library, because it is based on the library originally designed in Liu's laboratory at Harvard University.10 The Harvard library consists of a 60 base length variable internal region flanked on either side by 20 base length constant primer regions total of 100 bases. The final form of the library is 5′CTCCTCTGACTGTAACCACG787878786666787878787866678787878786666787878787866678787878GCATAGGTAGTCCAGAAGCC3′ where 6 is a mixture that produces 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 A/C/G/T, 7 is a mixture that produces 45:5:45:5 A/C/G/T, and 8 is a mixture that produces 5:45:5:45 A/C/G/T. After washing we eluted the bound aptamers and then incubated them with the CD20−HEK cells, this counter selection eliminated non-CD20 aptamers from the pool. After each round of selection the pool was gel-purified, PCR amplified and digested using lambda exonuclease in preparation for the next round. In total, 10 rounds of selection were performed with progressive increase in stringency to favour higher affinity pools to CD20+ cells (Table S1†). We evaluated the enrichment of pools to CD20+HEK cells using flow cytometry. The final aptamer pool from the round 10 exhibited statistically significant and specific binding with CD20+HEK cells as well as with the naturally CD20 expressing CCL-86 cells, but not with a naturally CD20 negative TIB-152 cells (Fig. S3†).
:1:1:1 A/C/G/T, 7 is a mixture that produces 45:5:45:5 A/C/G/T, and 8 is a mixture that produces 5:45:5:45 A/C/G/T. After washing we eluted the bound aptamers and then incubated them with the CD20−HEK cells, this counter selection eliminated non-CD20 aptamers from the pool. After each round of selection the pool was gel-purified, PCR amplified and digested using lambda exonuclease in preparation for the next round. In total, 10 rounds of selection were performed with progressive increase in stringency to favour higher affinity pools to CD20+ cells (Table S1†). We evaluated the enrichment of pools to CD20+HEK cells using flow cytometry. The final aptamer pool from the round 10 exhibited statistically significant and specific binding with CD20+HEK cells as well as with the naturally CD20 expressing CCL-86 cells, but not with a naturally CD20 negative TIB-152 cells (Fig. S3†).
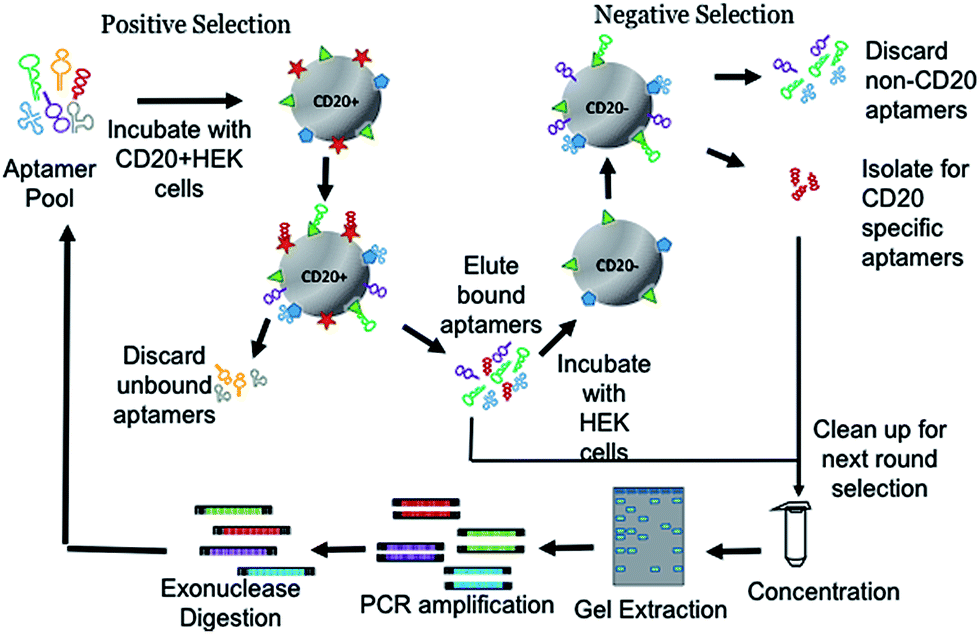 | ||
| Fig. 1 Generation of anti-CD20 aptamers with differential cell-SELEX. Lentiviral transfection of HEK cells generates target positive CD20+HEK cells. During positive selection aptamers that associate with the CD20+HEK cells are retained, while negative selection eliminates non-CD20 aptamer sequences. Each cycle culminates in a preparatory clean-up, which concentrates, purifies and amplifies the pool for the next round of selection. | ||
Pool 10 was sequenced by Eurofins Genomics Company using Illumina next-generation sequencing (NGS) technology, sequencing-by-synthesis, and the data were quantified and analysed using the Galaxy module.11 The results of NGS showed that pool 10 exhibited significant sequence convergence, with substantial decreases in the overall sequence complexity as well as the possession of high copy-number sequences, which prevailed in the pool (Fig. S4 and S5†). Once the NGS data were consolidated, the lead aptamer candidates, named NLA aptamer clones, were chemically synthesized by IDT DNA Technology and evaluated for specificity and affinity using flow cytometry. The NLA aptamers exhibited strong binding affinity with apparent Kds less than 75 nM, as well as high specificity to the CD20+HEK cells (Fig. 2A and S6†). The NLA aptamers showed also good binding to the naturally CD20 positive CCL-86 cells, but not that naturally CD20 negative TIB-152, this association was statistically significantly when compared to the synthetic DNA library as a nonspecific control (Fig. S7†).
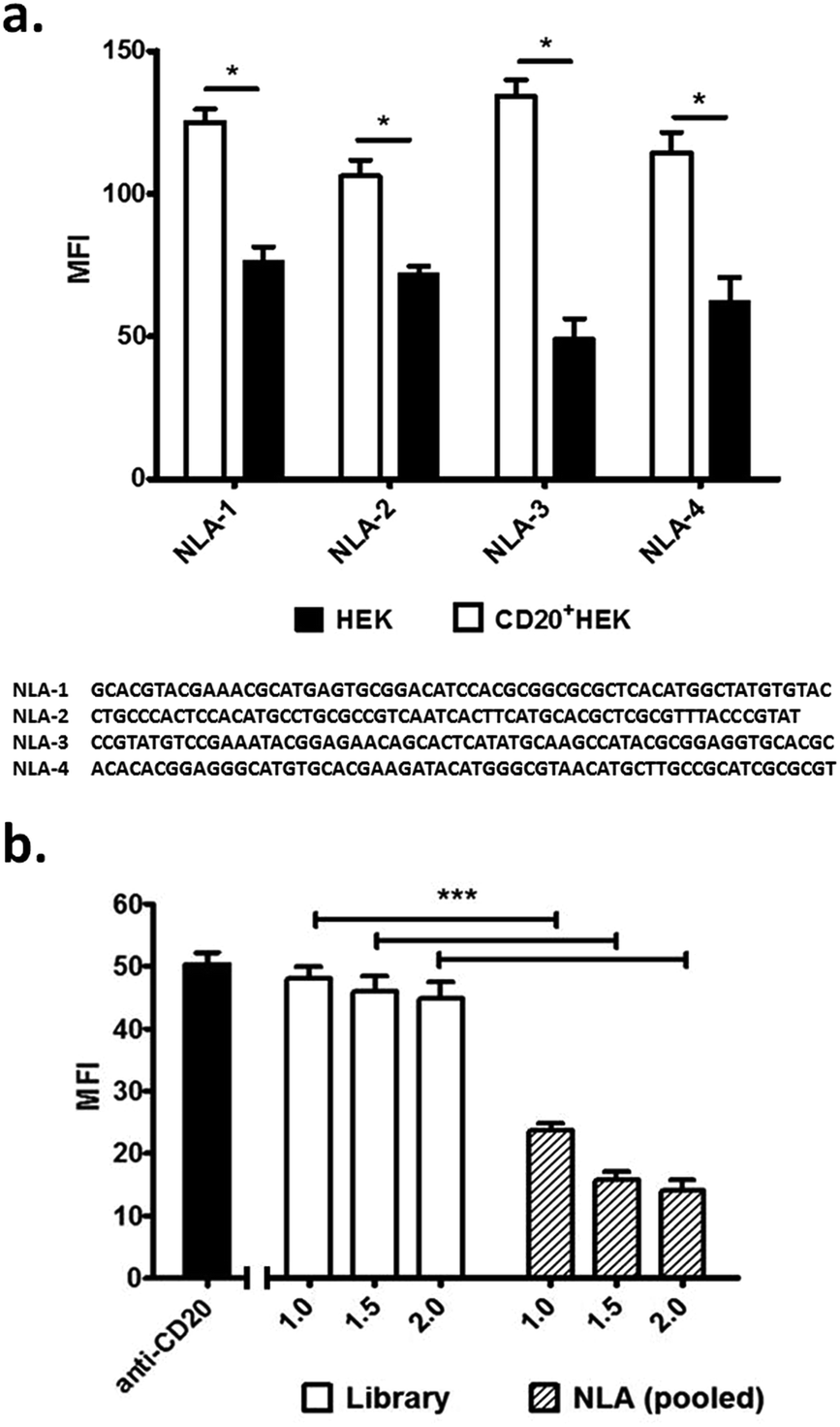 | ||
| Fig. 2 (a) NLA aptamer clones (400 nM) binding to CD20+HEK and HEK cells measured by flow cytometry. NLA sequences are shown without PCR primer sites. (b) The pre-incubation of CD20+HEK cells with pooled NLA aptamers leads to a total decrease of the anti-CD20 mAb binding (10 ng μL−1), as compared alongside with the anti-CD20 mAb only (solid black bar). Results above are of triplicate samples. MFI, median fluorescent intensity. *p < 0.05. | ||
To better assess the specificity of the NLA aptamers, we performed a competitive experiment using a commercially available anti-CD20 monoclonal antibody. For this the CD20+HEK cells were first incubated with either the DNA library or an equimolar amount of pooled NLA aptamers, and then subsequently stained with 10 ng μL−1 of anti-CD20 mAb. The extent of antibody binding was compared to a singly stained mAb control (Fig. 2B). Incubating CD20+HEK cells with 1 μM or more of pooled NLA aptamers dramatically decreased the antibody signal from the Median Fluorescent Intensity (MFI) of 55 to MFI of 22, with greater reductions at higher NLA concentrations. The use of the DNA library, however, showed no significant effect. This suggests that NLA aptamers have the capacity to act as physical antagonists shielding CD20 from antibody recognition.
To define the functionality of the NLA aptamers, we performed a CDC assay with intact and heat-inactivated human serum.4f CDC has an absolute requirement for various complement factors (C3b, C5 etc.), which are readily found in viable human serum. Heating human serum deactivates compliment factors and decreases apoptosis and necrosis of antibody-bound cells. Cellular viability was evaluated by measuring the signal intensity of the fluorescent vital stain 7-aminoactinomycin D (7-AAD) as well as the pro-apoptosis marker Annexin-V using flow cytometry. As can be seen from Fig. 3 and S8,† CDC activation and subsequent cell death was readily observed in the CD20+CCL-86 cells treated together with anti-CD20 mAb and viable human serum (a CDC positive control). The CDC caused a dramatic increase of 7-AAD and Annexin-V staining. Flow cytometry histograms revealed two distinct populations for 7-AAD staining; the population with the lower fluorescence intensity represents the living cells, whose membranes are still capable of excluding 7-AAD while the population of higher fluorescence intensity, representing 65% of cells after 4 hours, depicts the total amount of dead cells. There is also a significant elevation in Annexin-V staining from an MFI of 2 to an MFI of 64.
 | ||
| Fig. 3 Complement-dependent cytotoxicity (CDC) was induced using 10 ng μL−1 of anti-CD20 mAb and viable human serum. The NLA-protected CCL-86 cells exhibit greater overall cellular viability compared to the DNA library (Lib), as indicated by total 7-AAD staining as well as decreased staining of the pro-apoptotic marker Annexin-V. The histograms above represent one result of triplicate samples. | ||
To observe the effect of NLA aptamers on CDC, CCL-86 cells were pre-incubated with 2 μM of either a combination of the DNA library (CDC + Lib) or the NLA aptamers (CDC + NLA). Cells incubated with the DNA library exhibited no significant changes from the CDC positive control; however the NLA-treated cells exhibited statistically significant decreases of CDC induced cellular damage. The NLA-treated cells are more viable after CDC induction, with 41% exhibiting low 7-AAD staining compared to the 27% in the CDC positive control. The NLA-treated cells also exhibit significantly less Annexin-V staining, an MFI of 46. Together this demonstrates that NLA aptamers have a protective capacity, and by binding to the CD20 receptor are able to limit CDC.
To conclude, we selected DNA aptamers to CD20, a phosphoprotein on the surface of B lymphocytes and playing a major role in the regulation, activation, proliferation, and differentiation of these cells. Binding of antibodies to CD20+ cells leads to their death via complement-dependent cellular cytotoxicity or antibody-dependent cellular cytotoxicity. Our NLA aptamers were potent and specific binders of the CD20 molecule, on both the transfected CD20+HEK, and the naturally CD20 positive, CCL-86. By binding CD20, the NLA aptamers were also effective shielding agents, significantly reducing the binding capacity of anti-CD20 antibody. Biologically, this feature also made the NLA aptamers effective CDC-antagonists, resulting in 10% greater viability and a marked reduction in the staining of the pro-apoptosis marker Annexin-V.
The important feature of the differential cell-SELEX technology is that it produces synthetic affinity probes (aptamers) to cell surface receptors in their native state and conformation. Such aptamers can be directly used for a number of other applications. They can be utilized for cell isolation, cell visualization, and tracking cells in vivo. They can also be used to modulate activities of cell receptors and deliver different agents (e.g. siRNA and drugs) into the cells.
References
- T. F. Tedder and P. Engel, Immunol. Today, 1994, 15, 450–454 CrossRef CAS PubMed.
- (a) P. McLaughlin, A. J. Grillo-López, B. K. Link, R. Levy, M. S. Czuczman, M. E. Williams, M. R. Heyman, I. Bence-Bruckler, C. A. White and F. Cabanillas, J. Clin. Oncol., 1998, 16, 2825–2833 Search PubMed; (b) B. Coiffier, Oncogene, 2007, 26, 3603–3613 CrossRef CAS PubMed; (c) M. J. Glennie, R. R. French, M. S. Cragg and R. P. Taylor, Mol. Immunol., 2007, 44, 3823–3837 CrossRef CAS PubMed; (d) K. M. King and A. Younes, Expert Rev. Anticancer Ther., 2001, 1, 177–186 CrossRef CAS PubMed.
- (a) J. Golay, L. Zaffaroni, T. Vaccari, M. Lazzari, G.-M. Borleri, S. Bernasconi, F. Tedesco, A. Rambaldi and M. Introna, Blood, 2000, 95, 3900–3908 CAS; (b) A. D. Kennedy, M. D. Solga, T. A. Schuman, A. W. Chi, M. A. Lindorfer, W. M. Sutherland, P. L. Foley and R. P. Taylor, Blood, 2003, 101, 1071–1079 CrossRef CAS PubMed; (c) A. D. Kennedy, P. V. Beum, M. D. Solga, D. J. DiLillo, M. A. Lindorfer, C. E. Hess, J. J. Densmore, M. E. Williams and R. P. Taylor, J. Immunol., 2004, 172, 3280–3288 CrossRef CAS PubMed; (d) C. S. Zent, C. R. Secreto, B. R. LaPlant, N. D. Bone, T. G. Call, T. D. Shanafelt, D. F. Jelinek, R. C. Tschumper and N. E. Kay, Leuk. Res., 2008, 32, 1849–1856 CrossRef CAS PubMed.
- (a) M. J. Walport, N. Engl. J. Med., 2001, 344, 1058–1066 CrossRef CAS PubMed; (b) M. J. Walport, N. Engl. J. Med., 2001, 344, 1140–1144 CrossRef CAS PubMed; (c) M. C. Carroll, Nat. Immunol., 2004, 5, 981–986 CrossRef CAS PubMed; (d) D. Ricklin and J. D. Lambris, Nat. Biotechnol., 2007, 25, 1265–1275 CrossRef CAS PubMed; (e) J. V. Sarma and P. A. Ward, Cell Tissue Res., 2011, 343, 227–235 CrossRef CAS PubMed; (f) A. W. Pawluczkowycz, F. J. Beurskens, P. V. Beum, M. A. Lindorfer, J. G. van de Winkel, P. W. Parren and R. P. Taylor, J. Immunol., 2009, 183, 749–758 CrossRef CAS PubMed.
- D. Ricklin, G. Hajishengallis, K. Yang and J. D. Lambris, Nat. Immunol., 2010, 11, 785–797 CrossRef CAS PubMed.
- H. J. Muller-Eberhard, Annu. Rev. Immunol., 1986, 4, 503–528 CrossRef CAS PubMed.
- O. Bohana-Kashtan, L. Ziporen, N. Donin, S. Kraus and Z. Fishelson, Mol. Immunol., 2004, 41, 583–597 CrossRef CAS PubMed.
- (a) J. Papadimitriou, C. Drachenberg, M. Shin and B. Trump, Virchows Arch., 1994, 424, 677–685 CrossRef CAS PubMed; (b) Z. Fishelson, G. Attali and D. Mevorach, Mol. Immunol., 2001, 38, 207–219 CrossRef CAS PubMed.
- D. A. Daniels, H. Chen, B. J. Hicke, K. M. Swiderek and L. Gold, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15416–15421 CrossRef CAS PubMed.
- K. M. Ruff, T. M. Snyder and D. R. Liu, J. Am. Chem. Soc., 2010, 132, 9453–9464 CrossRef CAS PubMed.
- (a) B. Giardine, C. Riemer, R. C. Hardison, R. Burhans, L. Elnitski, P. Shah, Y. Zhang, D. Blankenberg, I. Albert and J. Taylor, Genome Res., 2005, 15, 1451–1455 CrossRef CAS PubMed; (b) D. Blankenberg, G. V. Kuster, N. Coraor, G. Ananda, R. Lazarus, M. Mangan, A. Nekrutenko and J. Taylor, Current protocols in molecular biology, 2010, 19.10. 11–19.10. 21 Search PubMed.
Footnotes |
| † Funding for this work was provided by NSERC Canada in the form of Discovery Grants to M. V. B. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra27165g |
| This journal is © The Royal Society of Chemistry 2016 |