
Asymmetric supercapacitor based on VS2 nanosheets and activated carbon materials
Tshifhiwa M. Masikhwa,
Farshad Barzegar,
Julien K. Dangbegnon,
Abdulhakeem Bello,
Moshawe J. Madito,
Damilola Momodu and
Ncholu Manyala*
Department of Physics, Institute of Applied Materials, SARCHI Chair in Carbon Technology and Materials, University of Pretoria, Pretoria 0028, South Africa. E-mail: ncholu.manyala@up.ac.za; Fax: +27 (0)12 420 2516; Tel: +27 (0)12 420 3549
First published on 1st April 2016
Abstract
An asymmetric supercapacitor was fabricated using VS2 nanosheets as the positive electrode and activated carbon (AC) as the negative electrode, with a 6 M KOH solution as electrolyte. These materials were combined to maximize the specific capacitance and enlarge the potential window, therefore improving the energy density of the device. A specific capacitance of 155 F g−1 at 1 A g−1 with a maximum energy density as high as 42 W h kg−1 and a power density of 700 W kg−1 was obtained for the asymmetric supercapacitor within the voltage range of 0–1.4 V. The supercapacitor also exhibited good stability, with ∼99% capacitance retention and no capacitance loss after 5000 cycles at a current density of 2 A g−1.
Introduction
Recently, supercapacitors, which are advanced electrochemical energy storage systems, have been a hot topic due to their numerous advantages in power source applications such as short-term power sources for mobile electronic devices and auxiliary power sources for hybrid electric vehicles (HEV).1–3 Supercapacitors, also known as ultracapacitors or electrochemical capacitors, are energy storage devices that exhibit high power density but relatively low energy density as compared to rechargeable batteries. Thus, there has been a massive focus by researchers to improve on supercapacitor technology in order to enhance the energy density to at least the level of rechargeable battery systems. Besides high power density, supercapacitors also show excellent properties such as long cycle life, rapid energy delivery and long stability as compared to rechargeable batteries. The performance of supercapacitors depends, to a large extent, on the nature of the electrode material.4,5Typically, supercapacitors are categorized into three types based on their charge-storage mechanism, namely, the electrical double-layer capacitors (EDLCs) and the faradaic and hybrid capacitors.2 The storage mechanism in EDLCs arises from the accumulation of charges at the electrode/electrolyte interface that results in a non-faradaic reaction, whereas the faradaic storage mechanism results from the fast redox reaction (faradaic reaction).6
Hybrid capacitors are obtained from either making composites of two materials with different charge storage mechanisms, or using the two materials as positive and negative electrodes to form an asymmetric cell. An example could be the use of an EDLC-type material as negative electrode and a faradaic-type material as positive electrode. This offers the possibility of synergizing the advantages of both electrodes, such as the high electrical conductivity and stability of EDLC materials and the high specific capacitance of faradic or pseudo-capacitance materials. The properties of the resulting hybrid device lie between those of a supercapacitor and a battery.7 Asymmetric supercapacitors are a special type of hybrid capacitors obtained by mounting an EDLC carbon material electrode with a faradaic-type material electrode in order to increase the energy density by utilizing the resulting wide operating voltage window. This should not be detrimental to the cyclic stability attributed to carbon-based materials and the excellent power density from the higher capacitance of the faradaic-type material.7–10
Carbon-based nanomaterials, such as activated carbon (AC), carbon nanotubes (CNTs), and graphene,11–15 are frequently used as the negative electrodes of asymmetric supercapacitors due to their stability in the negative potential region, good electronic conductivity, large surface area and relatively low cost.16–18 Activated carbon (AC) is the most suitable negative electrode material that has been adopted for hybrid capacitors, not only due to the numerous merits listed above but also its facile preparation process.19,20
Various transition metal oxides and conductive polymers21–25 are mostly used as positive electrodes due to their fast and reversible electron-exchange reactions at the electrode interface, which contribute to the high power densities and high capacitance of asymmetric supercapacitors.26–34
In recent years, metal chalcogenide materials have been considered for electrochemical energy storage applications due to their diverse chemical and physical properties.35 For example, two-dimensional (2D) layered transition-metal dichalcogenides (TMDs) such as MoS2, CoS2, VS2 and NiS2 have been extensively studied for applications in electrochemical supercapacitors due to their promising electrochemical performance.36–38 Numerous studies involving vanadium sulfides have been reported for various applications.37,39–42 Pandurangan et al. reported the synthesis of VS2/rGO nanosheets by an rGO-assisted phase transformation via annealing of VS4/rGO sheets at 350 °C for the electrocatalysis of hydrogen evolution reaction.43 Similarly, Satyajit et al. also provided a detailed study of VS4/rGO sheets synthesized by a facile, one-step hydrothermal route and with the enhanced supercapacitor performance of VS4/rGO.39 The experimental results were further confirmed by computational simulations to further provide useful insights into the design of efficient energy storage devices. However, there have been little or no reports on the fabrication of an asymmetric capacitor with activated carbon from polymer-based materials as the anode and VS2 as the cathode in an aqueous electrolyte.
In this work, we report on the design of an asymmetric capacitor based on 3D interconnected activated carbon as negative electrode and mesoporous VS2 nanosheets as positive electrode material. The hybrid material showed high rate capability compared with a pure VS2 nanosheet electrode. The VS2//AC asymmetric supercapacitor performed reversibly at a high cell voltage of 1.4 V in 6 M KOH. It exhibited a specific capacitance of 155 F g−1 at 1 A g−1 with a maximum energy density as high as 42 W h kg−1 and a power density of 700 W kg−1. Furthermore, the supercapacitor also exhibited good stability with ∼99% capacitance retention and no capacitance loss after 5000 cycles at a current density of 2 A g−1.
Experimental
Materials
Ammonium vanadate (NH4VO3, purity > 99.99%), thioacetamide (CH3CSNH2, Sigma-Aldrich, ≥ 99%), ammonia (NH3, ≥ 99.95%) and polyvinyl alcohol (PVA, 99+% hydrolyzed) were purchased from Sigma-Aldrich. Nickel foam was purchased from Alantum (Munich, Germany); potassium hydroxide (KOH, min 85%) and urea (purity ≥ 98%) were purchased from Merck (South Africa).Synthesis of VS2 nanosheets
Scheme 1 shows the schematic method used to prepare the VS2 nanosheets in this study. Ammonium metavanadate (NH4VO3, 2 mmol) was added to an 18 ml mixture of 15 ml deionized water and 3 ml ammonia, accompanied by vigorous stirring to completely dissolve the ammonium vanadate. Subsequently, thioacetamide (CH3CSNH2, 10 mmol) was added to the above solution under vigorous stirring. The final homogeneous solution was transferred into a sealed, Teflon-lined, stainless-steel autoclave and kept at a temperature of 180 °C for 20 h. After cooling to room temperature, the black powder was washed with deionized water and dried at 60 °C. The formation of the nanosheet morphology of the VS2 electrode includes two steps: an initial nucleating stage and a crystal growth stage including an Ostwald ripening process, which is known for the development of nanosheet metal sulfide structures.44 In the early stage, various functional groups present in the reaction vessel such as –NH2, –COOH, and –SH react with V4+ ions to form V–S complexes, which then decay to form VS2 nuclei in the last stage of the synthesis mechanism.45 In the second stage, nanosheet structures form as a result of the Ostwald ripening and self-assembly of the VS2 sheets. Throughout the reaction, the hydrolysis of thioacetamide produces HS−, which decreases V5+ to V4+, and layered VS2 structures are formed.46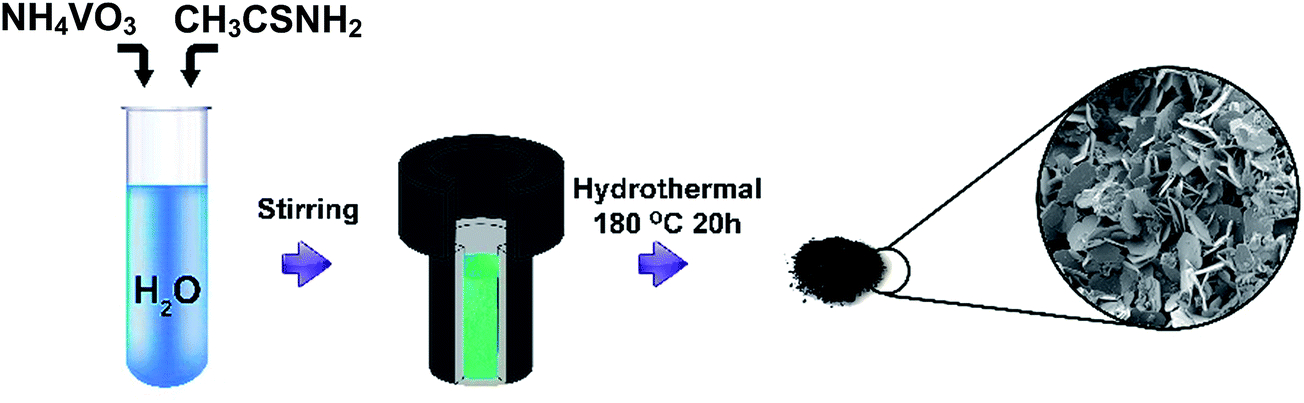 | ||
| Scheme 1 Preparation procedure of VS2 nanosheets. | ||
Synthesis of activated carbon
Activated carbon used for the production of the composite materials was prepared as reported in our earlier work.47,48 Briefly, graphene foam (GF) and polyvinyl alcohol (PVA) were used as starting material for the production of a hydrogel, which was then used to produce porous carbon materials after the activation process. The activated material was neutralized with 0.1 M HCl, washed with deionised water and dried at 120 °C for 12 h, after which samples were characterized.Structure and morphology characterization
The morphology of the prepared VS2 and activated carbon electrodes was studied using the high-resolution Zeiss Ultra Plus 55 field emission scanning electron microscope (FE-SEM) operated at 2.0 kV. Energy-dispersive X-ray (EDX) patterns were taken with a JEOL 5800LV microscope equipped with an energy-dispersive X-ray spectrometer operated at 20 kV, and they were used to estimate the elemental composition of the produced materials. Transmission electron microscopy (TEM) micro-images and selected area electron diffraction (SAED) observations were carried out with a JEOL JEM-2100F microscope operated at 200 kV (Akishima-shi, Japan). X-ray diffraction (XRD) patterns of the prepared materials were collected using an XPERT-PRO diffractometer (PANalytical BV, Netherlands) with theta/2theta geometry, operating with a cobalt tube at 50 kV and 30 mA and with reflection geometry at 2θ values ranging from 30–90° with a step size of 0.01°. The Raman spectra were recorded using a WITEC-Alpha 300R Plus confocal Raman spectrometer (WITEC GmbH, Ulm, Germany) with a 532 nm laser. X-ray photoelectron spectroscopy (XPS, K-alpha, Thermo Fisher) with monochromatic Al Kα radiation as the X-ray source was used to irradiate the sample surface and determine the chemistry of the samples synthesized in powder form. Nitrogen adsorption–desorption isotherms were measured at −196 °C using a Micromeritics ASAP 2020. All the samples were degassed at 180 °C for more than 12 h under vacuum conditions. The surface area was calculated by the Brunauer–Emmett–Teller (BET) method from the adsorption branch in the relative pressure range (P/P0) of 0.01–1.Electrode preparation and electrochemical characterization
All electrochemical measurements were carried out using a Biologic VMP-300 potentiostat (Knoxville, TN, USA) controlled by the EC-Lab® V10.37 software. Three-electrode measurements were performed for both AC and VS2 serving as the working electrodes, Ag/AgCl (3 M KCl) serving as the reference electrode and glassy carbon plate as the counter electrode. The negative electrode was prepared by mixing the active material of activated carbon (AC) with polyvinylidene fluoride (PVDF) binder in a weight ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which was then homogenized and dispersed in N-methylpyrrolidone (NMP) solution. The slurry was then uniformly pasted on a nickel foam current collector and dried at 60 °C in an oven for 8 h to ensure complete evaporation of the NMP. The positive electrode was prepared by mixing the active material (VS2), carbon black (CB) and polyvinylidene fluoride (PVDF) binder, which helped to improve the conductivity of material, in a weight ratio of 8:1:1 and then dispersing in N-methylpyrrolidone (NMP) solution. The slurry was then uniformly pasted on a nickel foam current collector and dried at 60 °C in an oven for 8 h.
:1, which was then homogenized and dispersed in N-methylpyrrolidone (NMP) solution. The slurry was then uniformly pasted on a nickel foam current collector and dried at 60 °C in an oven for 8 h to ensure complete evaporation of the NMP. The positive electrode was prepared by mixing the active material (VS2), carbon black (CB) and polyvinylidene fluoride (PVDF) binder, which helped to improve the conductivity of material, in a weight ratio of 8:1:1 and then dispersing in N-methylpyrrolidone (NMP) solution. The slurry was then uniformly pasted on a nickel foam current collector and dried at 60 °C in an oven for 8 h.
The electrochemical test of the asymmetric cell was carried out in a two-electrode cell configuration by means of coin-type cells with thickness of 0.2 mm and diameter of 16 mm, using a glass micro-fibre filter paper as the separator in a 6 M KOH aqueous electrolyte solution. The mass loading of VS2 (positive electrode) was within 2–3 mg and that of AC (negative electrode) within 6–9 mg.
Results and discussion
Fig. 1(a) shows the XRD patterns of the VS2 powder sample. The wavelength used for the XRD analysis was the 1.7890 Å line of a Co-Kα source. All diffraction peaks designated in the figure correspond to the pure hexagonal phase of VS2 (JCPDS 36-1139) with lattice constants of a = b = 0.322 Å and c = 0.576 Å. The XRD data suggest that VS2 preferentially grows in the (001) low-index crystallographic direction, hence leading to nanosheet formation. Fig. 1(b) presents the XRD patterns of AC powder. The wavelength used for the XRD analysis was Cu-Kα, 1.5405 Å. All the peaks are identified to those of graphite (COD: 96-900-8570), which crystallizes in the hexagonal structure with space group P63mc(186), and lattice parameters a = 2.4560 Å and c = 6.6960 Å.48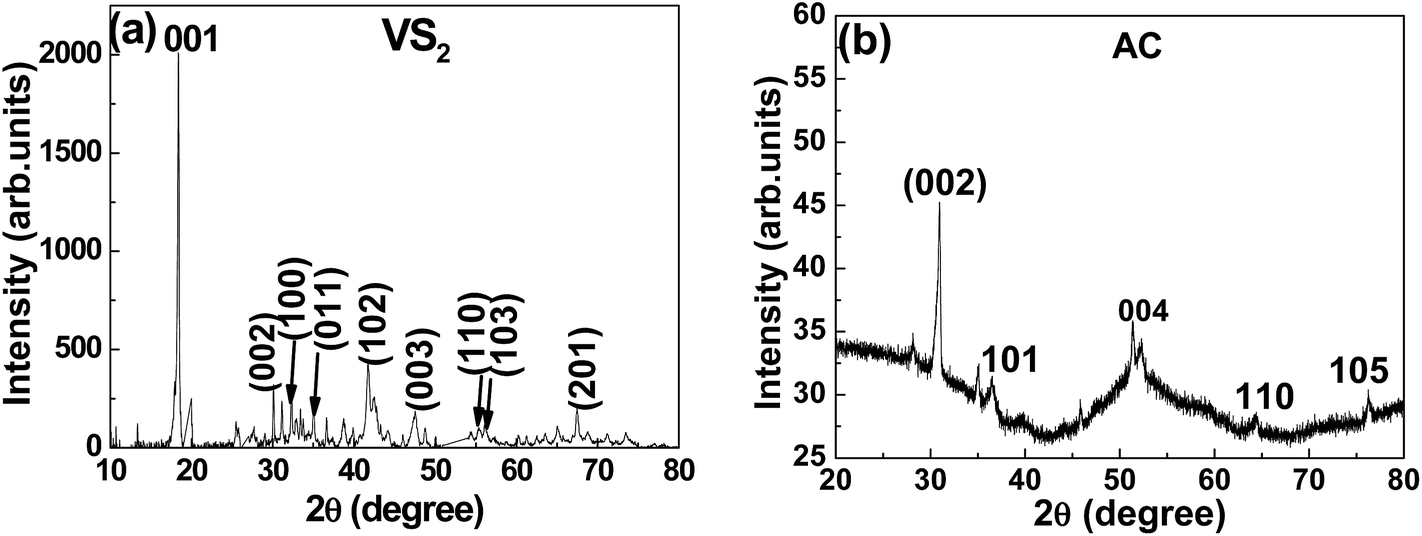 | ||
| Fig. 1 X-ray diffraction of (a) VS2 and (b) AC electrodes. | ||
Fig. 2(a) shows the Raman spectrum of the VS2 electrode. The characteristic peak positions of 278 cm−1 and 404 cm−1 due to the in-plane E12g and the out-of-plane A1g vibration modes show the presence of VS2.49 The A1g mode was due to the out-of-plane vibration of only the S atoms in opposite directions, while the in-plane E12g mode formed from the opposite vibration of the two S atoms with respect to the V atom.49
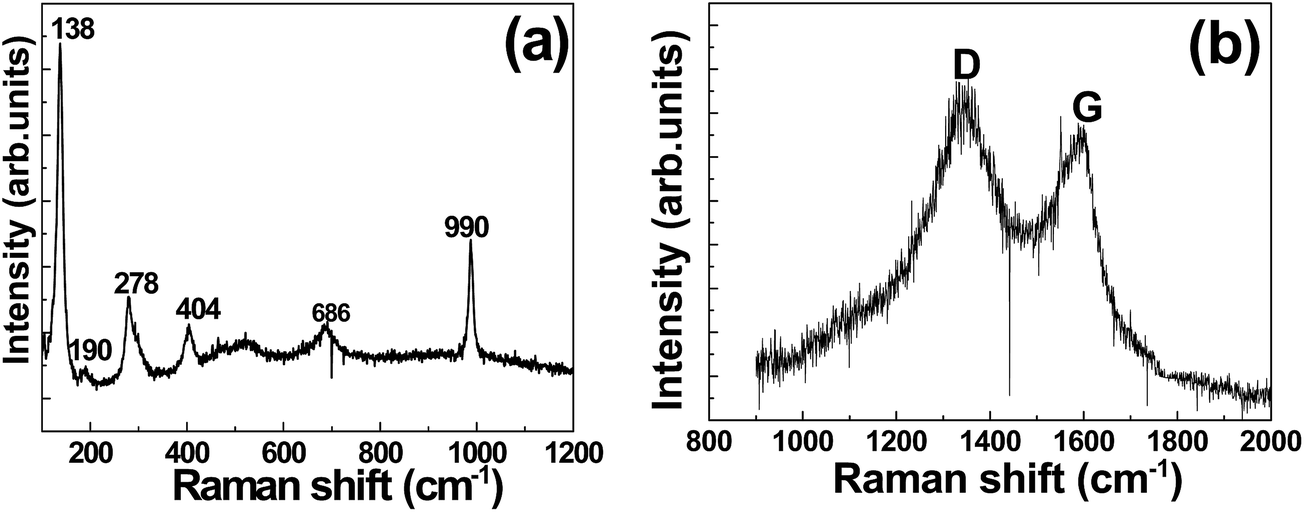 | ||
| Fig. 2 Raman pattern of (a) VS2 and (b) AC electrodes, respectively. | ||
Fig. 2(b) shows two prominent D and G peaks, signifying that the produced AC is composed of highly disordered graphitic structures. It shows two distinct peaks at 1344 cm−1 (D-band) and 1603 cm−1 (G-band). These bands correspond to the disordered carbon/structural defects and graphitic layers (sp2-bonded carbon atoms) of the carbon material.50 The intensity ratio of the D-band to that of G-band (R = ID/IG) is 0.8, and this shows a low degree of graphitic crystalline structure.51
Fig. 3(a) and (b) show the low- and high-magnification SEM micrographs of the VS2 sample. It can be seen from the figure that the sample is composed of a large number of nanosheets. Fig. 3(c) and (d) present the low and high magnification SEM of the AC sample. As observed in the micrograph, the sample is composed of large, interconnected macropores that provide a large ion-accessible surface for fast ion transport in high-performance supercapacitors.48
 | ||
| Fig. 3 Low- and high-magnification SEM images of (a) and (b) VS2, (c) and (d) AC electrodes. | ||
The high-resolution TEM (HRTEM) image of the VS2 nanosheets in Fig. 4(a) shows layered VS2 sheets with an interlayer spacing of 0.26 nm (upper inset to the figure), which corresponds to the (001) plane of the hexagonal phase of VS2. Fig. 4(b) shows the selected area electron diffraction (SAED) pattern of a single nanosheet, which is well indexed to the single-crystalline hexagonal phase of VS2. Fig. 4(c) shows the TEM image of activated carbon with large particles.
 | ||
| Fig. 4 (a) HRTEM image of VS2 nanosheets with the inset to the figure showing d spacing (upper), (b) SAED pattern of VS2 nanosheets, and (c) TEM image of the AC sample. | ||
The corresponding EDX pattern in Fig. 5(a) confirms that the synthesized VS2 composite is composed of the elements vanadium and sulfur in the sample.
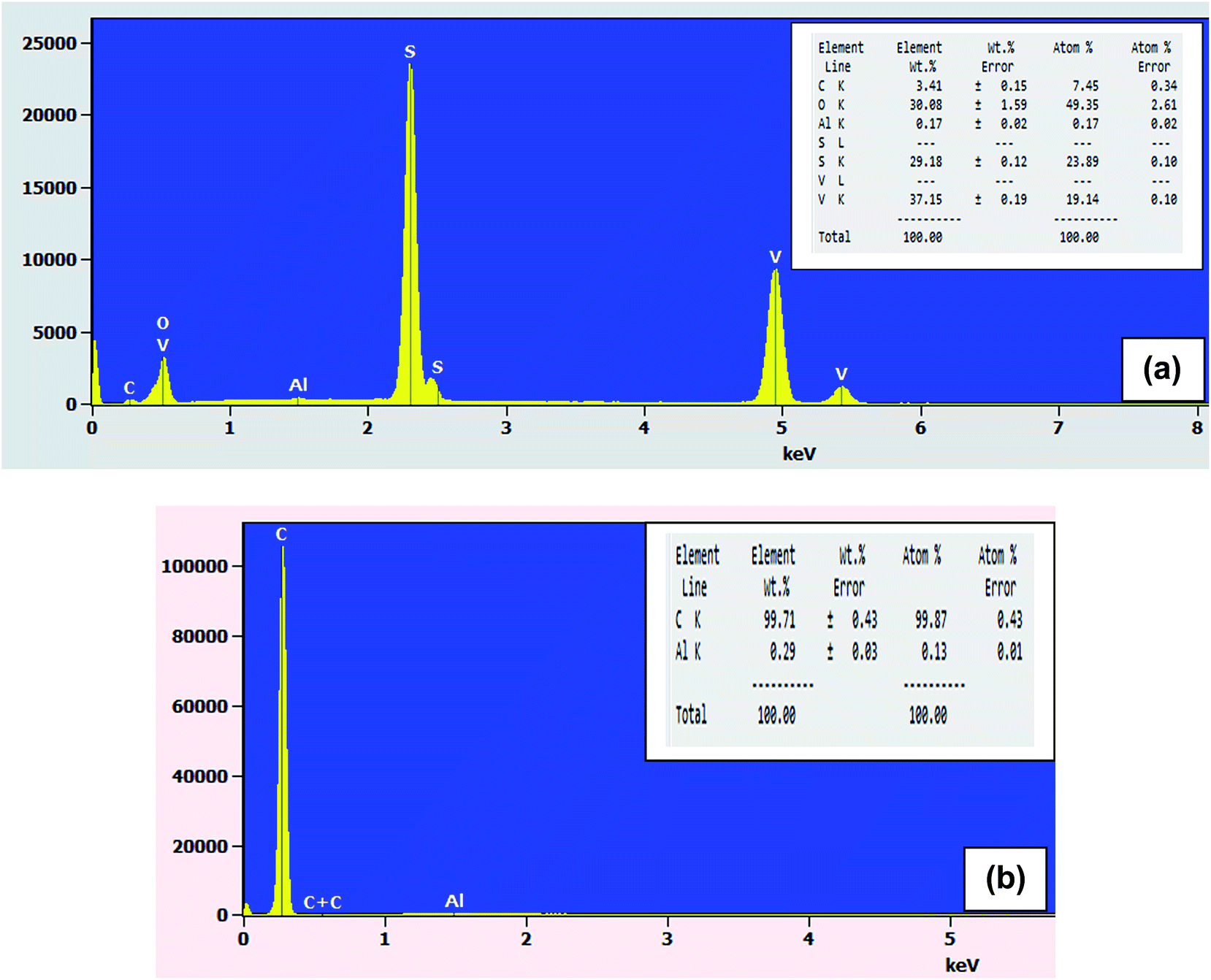 | ||
| Fig. 5 EDX pattern of (a) VS2 and (b) AC electrodes. | ||
Fig. 5(b) confirms that activated carbon is composed of carbon. The element Al is present due to signals from the copper grid used as a sample holder for microscopy.
The chemical composition of VS2 was also confirmed by XPS, as shown in Fig. 6. Before ion sputtering, C, O, V and S elements are detected in the survey; however, after ion sputtering for 30 minutes, only V and S elements are detected, which suggests that the appearance of C and O in the initial survey could be due to surface-adsorbed CO2 and O2 (see Fig. 6(a)). Fig. 6(b) and (c) show high-resolution scans of V 2p and S 2p, respectively. The energy positions of the V-peaks, which correspond to 513.5 eV and 521.1 eV (V 2p3/2 and V 2p1/2), indicate a V valence of +4.52,53 The S peaks corresponding to S 2p at 161.4 eV energy are comprised by S 2p1/2 (160.5 eV) and S 2p3/2 (161.9 eV) peaks, which are orbitals of divalent sulfide ions. Therefore, the survey after ion sputtering (showing only V and S elements) corresponds to a pure VS2 phase.
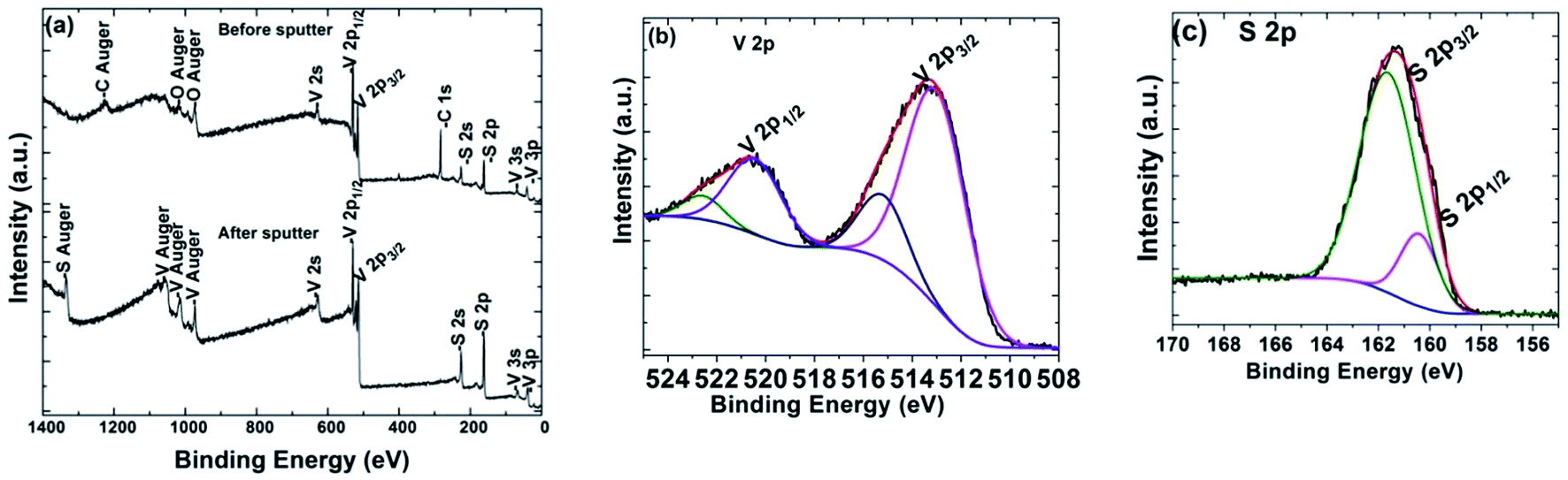 | ||
| Fig. 6 (a) VS2 XPS survey spectra and high-resolution scans of (b) V 2p and (c) S 2p. | ||
Fig. 7(a) and (b) represent the BET results from the surface area and porosimetry analysis carried out on the VS2 and the AC samples, respectively. The nitrogen adsorption–desorption isotherm of VS2 shows a type III behaviour with H3 hysteresis isotherm (Fig. 4(a)), indicating a weak interaction between the N2 adsorbent and the material. On the other hand, the N2 adsorption–desorption isotherm of the AC (Fig. 4(b)) presents a typical type I adsorption–desorption that characterizes complex materials containing micropores. The samples have a corresponding specific surface area (SSA) of 5.5 m2 g−1 and 1040 m2 g−1 for the VS2 and AC samples, respectively. In addition, the nature of the pore structure present in the VS2 nanosheets are mainly mesoporous with a broad pore size distribution of 2–7 nm (inset to Fig. 7(a)), while a microporous structure is recorded in the AC samples with an average pore size of 2.7 nm (inset to Fig. 7(b)).
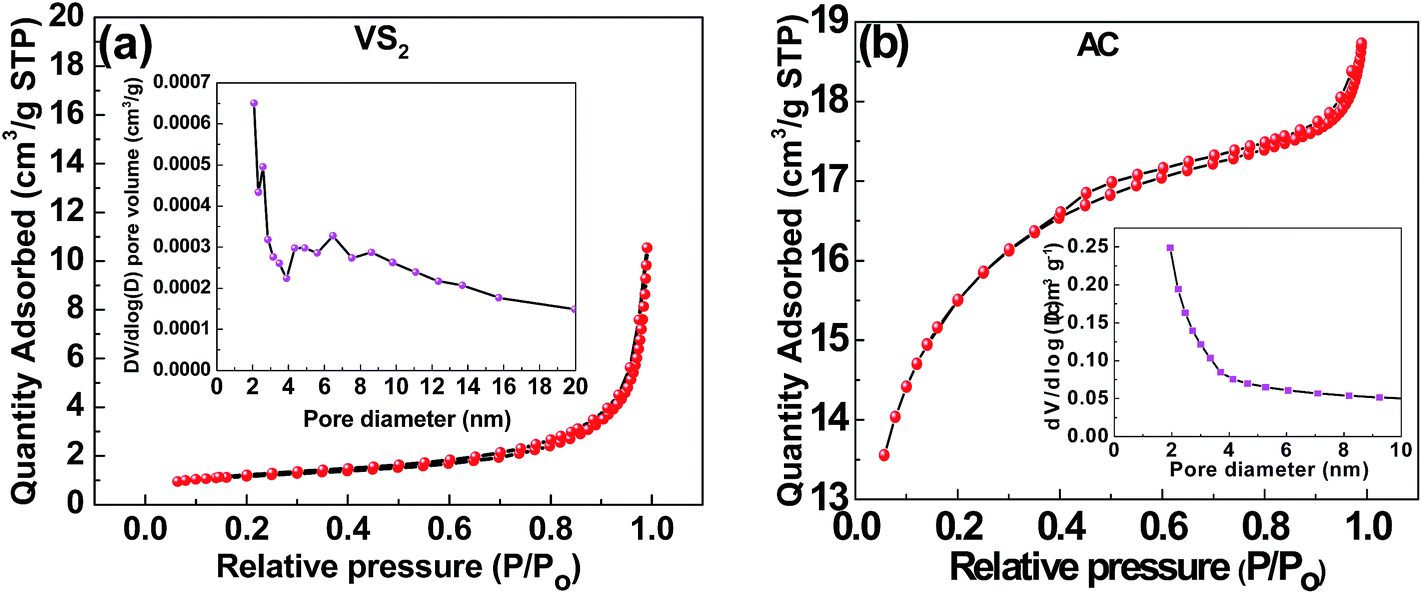 | ||
| Fig. 7 The N2 adsorption–desorption isotherm of the (a) VS2 nanosheets and (b) AC, insets show pore size distribution. | ||
Electrochemical performance of VS2 nanosheets and activated carbon
To evaluate the potential of the electrode materials for supercapacitor applications, the individual electrochemical properties of the VS2 and AC electrodes were firstly studied in a three-electrode system using 6 M KOH electrolyte. Cyclic voltammetry (CV), chronopotentiometry (CD) and electrochemical impedance spectroscopy (EIS) measurements were carried out for both samples. Fig. 8(a) shows the CV curves of the VS2 electrode at scan rates of 5, 10, 20, 50 and 100 mV s−1, respectively. A pair of redox peaks corresponding to the anodic peak at ∼0.25 V and cathodic peak at ∼0.34 V is visible in all the CV curves, revealing distinct faradic characteristics. Therefore, the anodic peak is due to the oxidation of V3+ to V4+, whereas the cathodic peak is due to the reverse process. With an increase in scan rates, the small shifts in both cathodic and anodic peak potentials are considered to have a direct relationship with the internal resistance of the electrode, indicating that the fast redox reactions occur at the electroactive material/electrolyte interface. Fig. 8(b) shows the CV curves of the AC electrode at various scan rates ranging from 5 to 100 mV s−1. These CV curves show relatively rectangular shapes without redox peaks, which is a typical electric double-layer capacitive sample behaviour.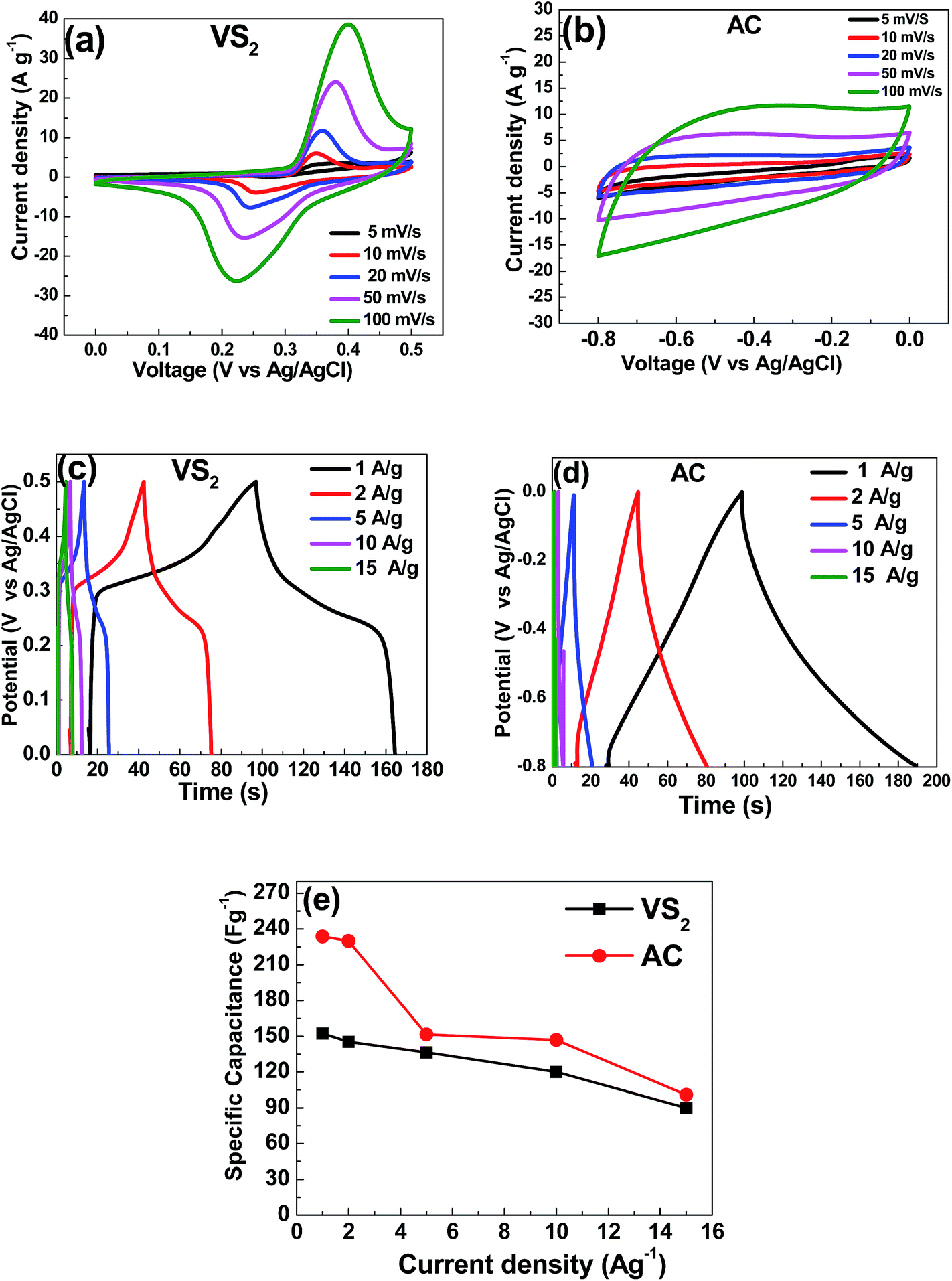 | ||
| Fig. 8 Cyclic voltammetry at scan rates of 5–100 mV s−1 for (a) VS2 and (b) AC electrodes; galvanostatic charge–discharge at current densities of 1–15 A g−1 for (c) VS2 and (d) AC electrodes; (e) specific capacitance versus current densities of the VS2 and AC electrodes. | ||
Fig. 8(c) shows the galvanostatic charge/discharge curves of the VS2 electrode at different current densities within a potential range of 0–0.5 V. Each discharge curve includes two clear voltage steps: a fast potential drop from 0.50 V to 0.35 V and a voltage plateau from 0.35 V to 0.22 V. The voltage plateau at around 0.3 V suggests a typical faradic characteristic that is in good agreement with the CV curves reported in Fig. 8(a). Fig. 8(d) shows the galvanostatic charge/discharge curves of the AC electrode at different current densities within a potential range of −0.8 to 0.0 V. As can be seen from Fig. 8(d), the shapes of the charge and discharge curves are very similar to each other and show a typical electric double-layer capacitive property, characterized by a nearly symmetrical triangular shape. This is in agreement with the results from CV curves in Fig. 8(b). Based on the galvanostatic charge–discharge (CD) curves, the specific capacitance of the VS2 and AC electrodes were calculated using eqn (1):
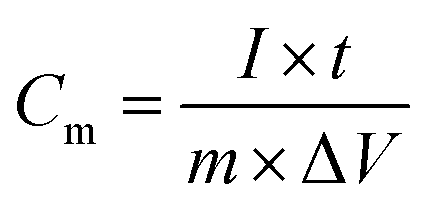 | (1) |
Electrochemical properties of the asymmetric supercapacitor
To further assess the potential application of the VS2 nanosheet array electrode in supercapacitors, an asymmetric supercapacitor was fabricated in which the positive electrode comprised of the VS2 material and the activated carbon material was used as the negative electrode. In order to obtain the optimal performance of the asymmetric full cell supercapacitor, a charge balance Q+ = Q− between the two electrodes was done, where Q+ and Q− are the charges stored in the positive and negative electrodes, respectively. The charge can be expressed by:54| Q = Cs × mΔU | (2) |
In order to get Q+ = Q−, the mass balancing follows the equation.
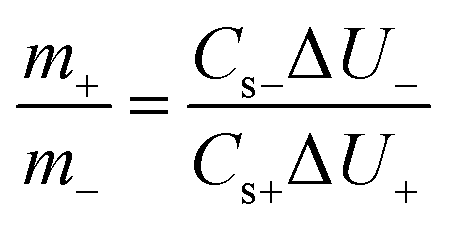 | (3) |
To make two-electrode cells, according to eqn (3), the mass of the VS2 electrode should be two times mass of the AC electrode.
From Fig. 9(a), it is expected that the operating cell voltage could be extended to about 1.4 V in 6 M KOH solution as electrolyte if the VS2 electrode as cathode and the AC electrode as anode are assembled into asymmetric ECs.
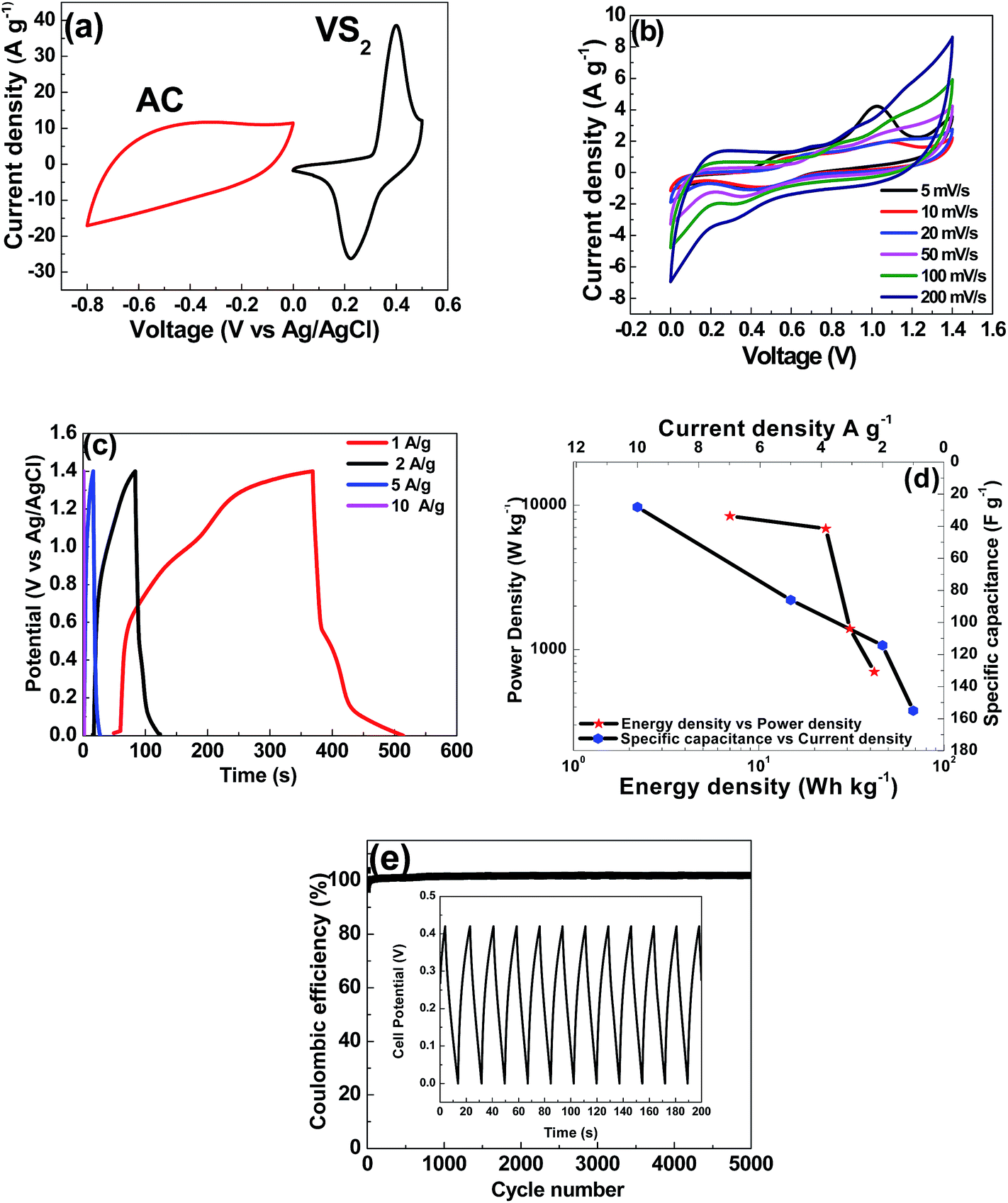 | ||
| Fig. 9 (a) Cyclic voltammetry of the VS2 nanosheet and activated carbon electrodes at a scan rate of 50 mV s−1 for the three-electrode setup. For the asymmetric VS2//AC device: (b) cyclic voltammetry at scan rates of 50–200 mV s−1, (c) galvanostatic charge–discharge at current densities of 1–10 A g−1, (d) Ragone plot and the specific capacitance as a function of the current density, and (e) cycle stability at a constant current density of 2 A g−1. | ||
Fig. 9(b) shows the CV curves of the VS2//AC asymmetric device measured at different scan rates from 5 to 200 mV s−1. It indicates that the CV curves simultaneously express the faradic and electric double-layer capacitive behaviour, which are the typical characteristics of hybrid asymmetric supercapacitors. An asymmetric charge–discharge curve was observed showing the existence of both faradic behaviour (0.55–0.15 V) and electric double-layer behaviour (0.55 V–1.4 V) at all current densities (Fig. 9(c)).
The energy density (E, in W h kg−1) and power density (P, in W kg−1) of ECs can be calculated from the specific capacitance, Cs according to the following equations:
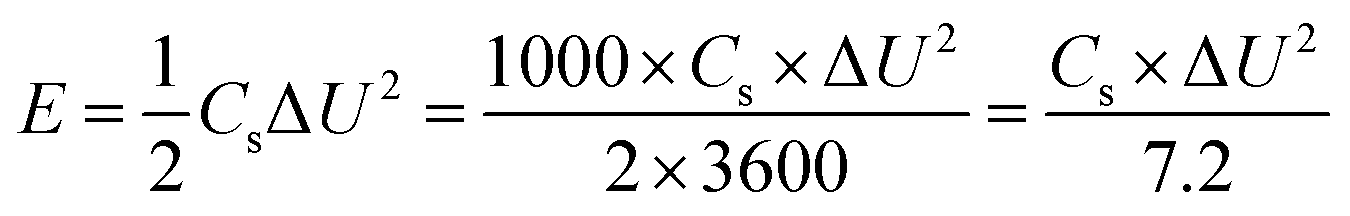 | (4) |
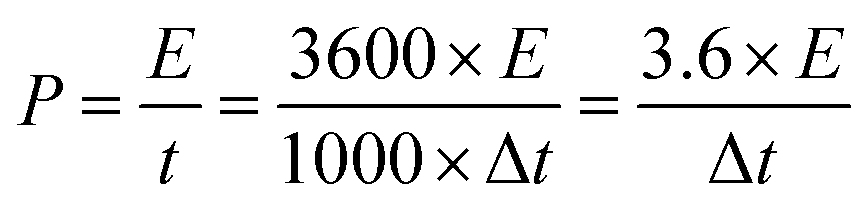 | (5) |
The Ragone plot and the specific capacitance as a function of the current density of the asymmetric device are shown in Fig. 9(d). The specific capacitance of the VS2//AC can reach 155 F g−1 at a current density of 1 A g−1 as calculated from eqn (2).
The maximum energy density of the device was recorded as 42 W h kg−1 with a corresponding power density of 700 W Kg−1 at a current density of 1 A g−1, as shown in Fig. 9(d). The asymmetric VS2//AC shows a much improved energy density at high power density in comparison with other asymmetric devices involving AC as negative electrode, such as AC//Ni(OH)2/XC-72 (energy density of 36 W h kg−1 and corresponding power density of 490 W kg−1 at 0.5 A g−1),55 Ni(OH)2@3D Ni-AC (energy density of 21.8 W h kg−1 and corresponding power density of 660 W kg−1 at 1 A g−1)56 and CuS//AC (energy density of 15.06 W h kg−1 and corresponding power density of 392.9 W kg−1 at 0.5 A g−1).57
The stability of the electrode material is also a very important characteristic for its application as energy storage device. In order to understand the stability of the VS2//AC asymmetric device, the samples were subjected to 5000 cycles at the high current density of 2 A g−1 as shown in Fig. 9(e). The coulombic efficiency of VS2//AC after 5000 cycles is ∼99%, thus exhibiting its excellent electrochemical stability.
Fig. 10(a) presents the Nyquist plot of the asymmetric device with an Rs value of 2.97 Ω obtained from the fitted plot using the circuit shown in the inset to the figure. The Nyquist plot before and after cycling is shown in Fig. 10(b). From this, an Rs-intercept value of 2.97 Ω before cycling and 4.10 Ω after cycling was obtained.
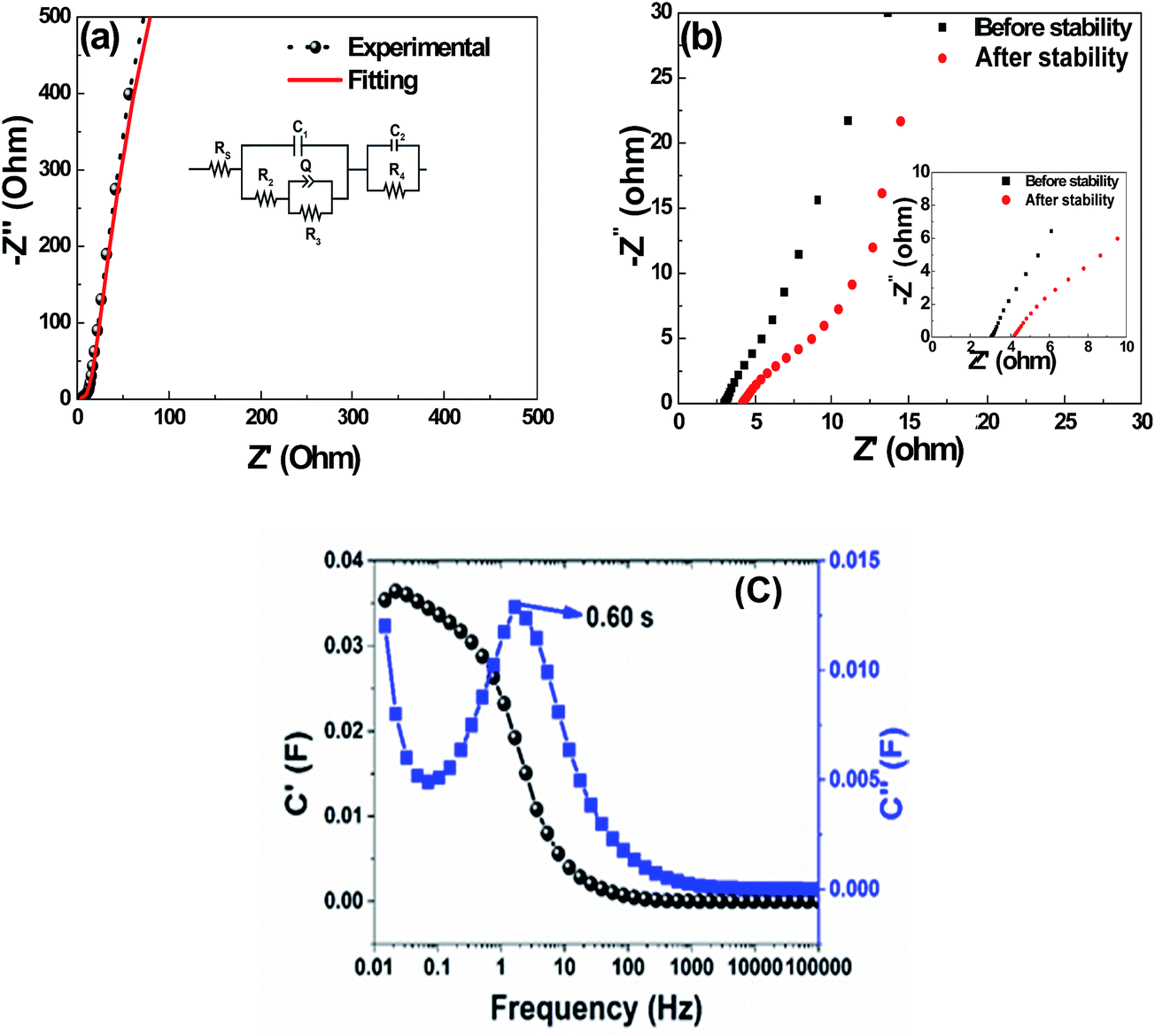 | ||
| Fig. 10 (a) EIS plot and fitting curve for the asymmetric VS2//AC cell, (b) EIS before and after cycling, (c) the real and the imaginary parts of the asymmetric cell's capacitance against frequency. | ||
The frequency response of porous carbon electrodes has been modelled with a single series resistor–capacitor (RC) circuit.58 Based on this model, the real and imaginary part of the capacitance as a function of the frequency can be calculated using the equations below:
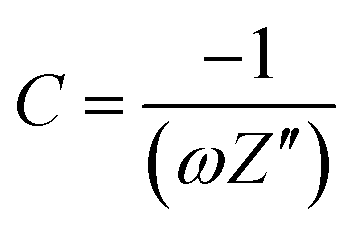 | (6) |
| C(ω) = C′(ω) − jC′′(ω) | (7) |
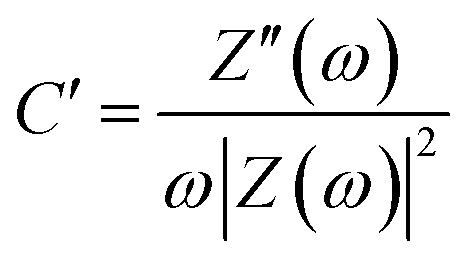 | (8) |
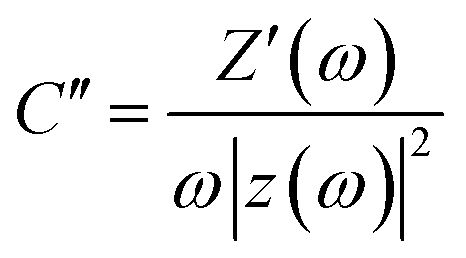 | (9) |
Conclusions
We have successfully fabricated an asymmetric supercapacitor cell based on porous activated carbon material as negative electrode and VS2 as positive electrode. The asymmetrical device displays a high specific capacitance of 155 F g−1 at 1 A g−1 with a maximum energy density of 42 W h kg−1 and power density of 700 W kg−1. In addition, its great stability record, with ∼99% capacitance retention and no capacitance loss after 5000 cycles at a current density of 2 A g−1 and an operating voltage of about 1.4 V in 6 M KOH aqueous electrolytes, shows that pairing such hybrid materials could be an excellent method to produce supercapacitors with high energy and power densities. These results offer a convenient and effective way to fabricate asymmetric hybrid supercapacitors based on VS2 and AC with high energy density while maintaining the high power density property of supercapacitors.Acknowledgements
This study is based on research supported by the South African Research Chairs Initiative of the Department of Science and Technology (SARChI-DST) and the National Research Foundation (NRF). Any opinions, findings and conclusions, or recommendations expressed in this study are those of authors, and therefore the NRF and SARChI-DST do not accept any liability with regard thereto. The financial support of University of Pretoria and NRF are gratefully acknowledged.References
- A. S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS PubMed.
- B. E. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, Kluwer Academic/Plenum, New York, 1999 Search PubMed.
- W. Van Schalkwijk and B. Scrosati, Advances in lithium-ion batteries, Springer Science & Business Media, 2002 Search PubMed.
- L. Zhang, F. Zhang, X. Yang, G. Long, Y. Wu, T. Zhang, K. Leng, Y. Huang, Y. Ma, A. Yu and Y. Chen, Sci. Rep., 2013, 3, 1408 Search PubMed.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- J. W. Long, B. Dunn, D. R. Rolison and H. S. White, Chem. Rev., 2004, 104, 4463–4492 CrossRef CAS PubMed.
- Y.-G. Wang, L. Cheng and Y.-Y. Xia, J. Power Sources, 2006, 153, 191–196 CrossRef CAS.
- S.-B. Ma, K.-W. Nam, W.-S. Yoon, X.-Q. Yang, K.-Y. Ahn, K.-H. Oh and K.-B. Kim, Electrochem. Commun., 2007, 9, 2807–2811 CrossRef CAS.
- V. Ganesh, S. Pitchumani and V. Lakshminarayanan, J. Power Sources, 2006, 158, 1523–1532 CrossRef CAS.
- H. Inoue, T. Morimoto and S. Nohara, Electrochem. Solid-State Lett., 2007, 10, A261–A263 CrossRef CAS.
- T.-W. Lin, C.-S. Dai, T.-T. Tasi, S.-W. Chou, J.-Y. Lin and H.-H. Shen, Chem. Eng. J., 2015, 279, 241–249 CrossRef CAS.
- W. Lin, W. Yu, Z. Hu, W. Ouyang, X. Shao, R. Li and D. S. Yuan, Electrochim. Acta, 2014, 143, 331–339 CrossRef CAS.
- T. Peng, H. Wang, H. Yi, Y. Jing, P. Sun and X. Wang, Electrochim. Acta, 2015, 176, 77–85 CrossRef CAS.
- J.-W. Lang, L.-B. Kong, M. Liu, Y.-C. Luo and L. Kang, J. Solid State Electrochem., 2010, 14, 1533–1539 CrossRef CAS.
- Q. T. Qu, Y. Shi, L. L. Li, W. L. Guo, Y. P. Wu, H. P. Zhang, S. Y. Guan and R. Holze, Electrochem. Commun., 2009, 11, 1325–1328 CrossRef CAS.
- D. Xuan, W. Chengyang, C. Mingming, J. Yang and W. Jin, J. Phys. Chem. C, 2009, 113, 2643–2646 Search PubMed.
- B. G. Choi, S.-J. Chang, H.-W. Kang, C. P. Park, H. J. Kim, W. H. Hong, S. Lee and Y. S. Huh, Nanoscale, 2012, 4, 4983–4988 RSC.
- P. Tang, L. Han and L. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 10506–10515 CAS.
- P. Simon and Y. Gogotsi, Acc. Chem. Res., 2012, 46, 1094–1103 CrossRef PubMed.
- Y.-G. Wang, Z.-D. Wang and Y.-Y. Xia, Electrochim. Acta, 2005, 50, 5641–5646 CrossRef CAS.
- M. Kumar, A. Subramania and K. Balakrishnan, Electrochim. Acta, 2014, 149, 152–158 CrossRef CAS.
- S. J. Zhu, J. Zhang, J. J. Ma, Y. X. Zhang and K. X. Yao, J. Power Sources, 2015, 278, 555–561 CrossRef CAS.
- Y. Xiao, Y. Cao, Y. Gong, A. Zhang, J. Zhao, S. Fang, D. Jia and F. Li, J. Power Sources, 2014, 246, 926–933 CrossRef CAS.
- H. R. Ghenaatian, M. F. Mousavi and M. S. Rahmanifar, Electrochim. Acta, 2012, 78, 212–222 CrossRef CAS.
- Y. Zhou, N. Lachman, M. Ghaffari, H. Xu, D. Bhattacharya, P. Fattahi, M. R. Abidian, S. Wu, K. K. Gleason and B. L. Wardle, J. Mater. Chem. A, 2014, 2, 9964–9969 CAS.
- M.-Y. Cho, S.-M. Park, J.-W. Lee and K.-C. Roh, J. Electrochem. Sci. Technol., 2011, 2, 152–156 CrossRef CAS.
- Y. Wang, L. Yu and Y. Xia, J. Electrochem. Soc., 2006, 153, A743–A748 CrossRef CAS.
- T. Brousse, P.-L. Taberna, O. Crosnier, R. Dugas, P. Guillemet, Y. Scudeller, F. Favier, Y. Zhou, P. Simon and D. Blanger, in Meeting Abstracts, 2006, p. 125 Search PubMed.
- L.-M. Chen, Q.-Y. Lai, Y.-J. Hao, Y. Zhao and X.-Y. Ji, J. Alloys Compd., 2009, 467, 465–471 CrossRef CAS.
- S. W. Lee, J. Kim, S. Chen, P. T. Hammond and Y. Shao-Horn, ACS Nano, 2010, 4, 3889–3896 CrossRef CAS PubMed.
- M. A. Bissett, I. A. Kinloch and R. A. W. Dryfe, ACS Appl. Mater. Interfaces, 2015, 7, 17388–17398 CAS.
- S. Nohara, T. Asahina, H. Wada, N. Furukawa, H. Inoue, N. Sugoh, H. Iwasaki and C. Iwakura, J. Power Sources, 2006, 157, 605–609 CrossRef CAS.
- D. P. Dubal, G. S. Gund, C. D. Lokhande and R. Holze, Energy Technol., 2014, 2, 401–408 CrossRef CAS.
- W. Tang, L. Liu, S. Tian, L. Li, Y. Yue, Y. Wu and K. Zhu, Chem. Commun., 2011, 47, 10058–10060 RSC.
- S. Surendran, K. V. Sankar, L. J. Berchmans and R. K. Selvan, Mater. Sci. Semicond. Process., 2015, 33, 16–23 CrossRef CAS.
- K. Krishnamoorthy, G. K. Veerasubramani, S. Radhakrishnan and S. J. Kim, Mater. Res. Bull., 2014, 50, 499–502 CrossRef CAS.
- J. Feng, X. Sun, C. Wu, L. Peng, C. Lin, S. Hu, J. Yang and Y. Xie, J. Am. Chem. Soc., 2011, 133, 17832–17838 CrossRef CAS PubMed.
- P. Justin and G. R. Rao, Int. J. Hydrogen Energy, 2010, 35, 9709–9715 CrossRef CAS.
- S. Ratha, S. R. Marri, N. A. Lanzillo, S. Moshkalev, S. K. Nayak, J. N. Behera and C. S. Rout, J. Mater. Chem. A, 2015, 3, 18874–18881 CAS.
- L. Li, S. Kim, W. Wang, M. Vijayakumar, Z. Nie, B. Chen, J. Zhang, G. Xia, J. Hu and G. Graff, Adv. Energy Mater., 2011, 1, 394–400 CrossRef CAS.
- H. A. Therese, F. Rocker, A. Reiber, J. Li, M. Stepputat, G. Glasser, U. Kolb and W. Tremel, Angew. Chem., Int. Ed., 2005, 44, 262–265 CrossRef CAS PubMed.
- J. Feng, L. Peng, C. Wu, X. Sun, S. Hu, C. Lin, J. Dai, J. Yang and Y. Xie, Adv. Mater., 2012, 24, 1969–1974 CrossRef CAS PubMed.
- P. Mohan, J. Yang, A. Jena and H. Suk Shin, J. Solid State Chem., 2015, 224, 82–87 CrossRef CAS.
- F. Cao, W. Hu, L. Zhou, W. Shi, S. Song, Y. Lei, S. Wang and H. Zhang, Dalton Trans., 2009, 9246–9252 RSC.
- C. Song, K. Yu, H. Yin, H. Fu, Z. Zhang, N. Zhang and Z. Zhu, J. Mater. Chem. C, 2014, 2, 4196–4202 RSC.
- C. S. Rout, R. Khare, R. V. Kashid, D. S. Joag, M. A. More, N. A. Lanzillo, M. Washington, S. K. Nayak and D. J. Late, Eur. J. Inorg. Chem., 2014, 2014, 5331–5336 CrossRef CAS.
- A. Bello, F. Barzegar, D. Momodu, J. Dangbegnon, F. Taghizadeh and N. Manyala, Electrochim. Acta, 2015, 151, 386–392 CrossRef CAS.
- F. Barzegar, A. Bello, D. Y. Momodu, J. K. Dangbegnon, F. Taghizadeh, M. J. Madito, T. M. Masikhwa and N. Manyala, RSC Adv., 2015, 5, 37462–37468 RSC.
- X. Liu, H.-L. Shuai and K.-J. Huang, Anal. Methods, 2015, 7, 8277–8284 RSC.
- G. Ma, D. Guo, K. Sun, H. Peng, Q. Yang, X. Zhou, X. Zhao and Z. Lei, RSC Adv., 2015, 5, 64704–64710 RSC.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed.
- J. Mendialdua, R. Casanova and Y. Barbaux, J. Electron Spectrosc. Relat. Phenom., 1995, 71, 249–261 CrossRef CAS.
- T.-H. Yang, S. Nori, H. Zhou and J. Narayan, Appl. Phys. Lett., 2009, 95, 2506 Search PubMed.
- A. Bello, F. Barzegar, D. Momodu, J. Dangbegnon, F. Taghizadeh, M. Fabiane and N. Manyala, J. Power Sources, 2015, 273, 305–311 CrossRef CAS.
- L. Sui, S. Tang, Z. Dai, Z. Zhu, H. Huangfu, X. Qin, Y. Deng and G. M. Haarberg, New J. Chem., 2015, 39, 9363–9371 RSC.
- Y.-Z. Su, K. Xiao, N. Li, Z.-Q. Liu and S.-Z. Qiao, J. Mater. Chem. A, 2014, 2, 13845–13853 CAS.
- J. Zhang, H. Feng, J. Yang, Q. Qin, H. Fan, C. Wei and W. Zheng, ACS Appl. Mater. Interfaces, 2015, 7, 21735–21744 CAS.
- P. L. Taberna, P. Simon and J.-F. Fauvarque, J. Electrochem. Soc., 2003, 150, A292–A300 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |