
Targeting CD44, ABCG2 and CD133 markers using aptamers: in silico analysis of CD133 extracellular domain 2 and its aptamer†
Abstract
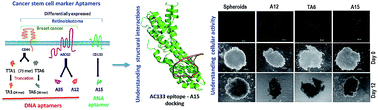
The application of nucleic acid aptamers for the diagnosis and therapy of cancer stem cells (CSCs) is expanding. The current study truncated and probed various existing aptamers against CSC markers CD44, ABCG2 and CD133 in retinoblastoma (RB) primary cells, cell lines, a breast cancer cell line and MCF7-sphere. Truncated CD44 aptamer retained its specific binding to cancer cells, ABCG2+ve MCF7-spheres and CD133+ve RB cells. Similarly, ABCG2 and CD133 aptamers showed higher affinity to ABCG2+ve, CD133+ve cells than the negative population and cell lines. All aptamers appreciably reduced viability of up to 50% and 32% of the primary RB tumor cells and cell lines, respectively. Colony formation of MCF7, RB cell lines and MCF7-sphere growth were inhibited significantly. Structure prediction, simulation of CD133 extracellular domain 2 (ExD2) and A15 followed by docking to comprehend the potential interaction revealed hydrogen bonds and non bonded interactions between them. This information could be used to improve the A15 aptamer to gain more interactions with CD133. Thus approaches undertaken here can be applied universally for cell-specific targeting, and the aptamers studied against CSC markers deserve further in vivo studies.
 Please wait while we load your content...
Please wait while we load your content...

