
Oxidative degradation of poly(3-hydroxybutyrate). A new method of synthesis for the malic acid copolymers†
 a
Piotr
Kurcok
*a
a
Piotr
Kurcok
*a
Abstract
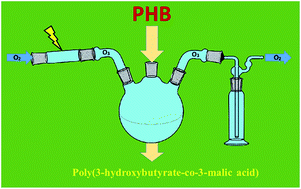
The thermal stability of poly(3-hydroxybutyrate) (PHB) in an oxidation environment was investigated in bulk at temperatures ranging from 100 °C to 140 °C. The process carried out in pure oxygen resulted in PHB backbone degradation with resulting non-volatile products typical for regular PHB thermal degradation while thermal treatment of PHB in an oxygen/ozone mixture resulted in increased rate of polymer backbone scission. The non-volatile degradation product contained macromolecules with several types of terminal groups but also a part of the 3-hydroxybutyrate repeating units was transformed into 3-malic acid units. NMR and multi-stage MS characterization revealed the random distribution of 3-malic acid units in the oligomeric products as well as the content of the malic acid units being dependent on oxidation conditions.
 Please wait while we load your content...
Please wait while we load your content...

