
Design and synthesis of potent N-phenylpyrimidine derivatives for the treatment of skin cancer†
Abstract
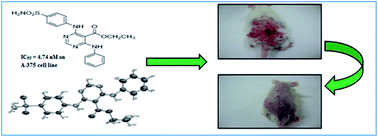
The development of novel synthetic compounds for the treatment of skin cancer is much needed, as there is a sudden rise in the incidence of skin cancer throughout the world and the available chemotherapy is facing problems of resistance. Hence, present research efforts have been made to discover potent molecules against skin cancer. Pharmacophore models were developed using the GALAHAD module of Sybyl X, followed by validation, virtual screening, design and in silico ADMET studies. Based on features generated in the computational studies and the structural importance of the pyrimidine moiety with the highest QFIT value molecule for virtual hits; it was selected as a core moiety for further designing molecules of interest. Fourteen substituted pyrimidine derivatives were designed, synthesized and characterised by 1H and 13C NMR, mass and elemental analysis, while the purity was checked by HPLC. All these compounds were evaluated in in vitro studies on five cancer lines; from which, four compounds were found to be potent, specifically with the skin cancer cell line. Based on the results of the in vitro studies, they were selected further, along with 5-FU, for an in vivo study with the DMBA-induced skin cancer model. One compound, 6h, showed favourable action against skin cancer and treated tumours, demonstrating the potential of the series. This study could be explored in future to design lead molecule for the treatment of skin cancer.
 Please wait while we load your content...
Please wait while we load your content...

