
Facile self-templating preparation of polyacrylonitrile-derived hierarchical porous carbon nanospheres for high-performance supercapacitors†
Long Yao*ab,
Guangzhi Yang*b and
Pan Hanab
aSchool of Environment and Architecture, University of Shanghai for Science and Technology, Shanghai 200093, China. E-mail: ylenv@sina.cn; Fax: +86-21-5527-0305; Tel: +86-21-5527-0305
bSchool of Materials Science and Engineering, University of Shanghai for Science and Technology, Shanghai 200093, China
First published on 26th April 2016
Abstract
A green, facile and efficient strategy was proposed to successfully synthesize polyacrylonitrile-derived hierarchical porous carbon nanospheres (HPCNs) for high-performance supercapacitors by surfactant-free emulsion polymerization followed with one-step KOH activation. The as-obtained HPCNs show favorable features for electrochemical energy storage such as a high specific surface area of up to 3130 m2 g−1, high volume of hierarchical pores up to 1.87 cm3 g−1, hierarchical porosity consisting of micro, meso, and macropores, turbostratic carbon structure, controlled and tunable pore size and stable thermal and chemical properties. The symmetric supercapacitor exhibits a reversible specific capacitance of 240 F g−1 at a current density of 1 A g−1 and displays a high energy density of 77 W h kg−1 at a power density of 875 W kg−1. A high specific capacitance retention of 96% could be maintained even after 3000 cycles. Moreover, we used different electrolytes to study the capacitive behavior with controlled pore size. The facile, efficient and template-free synthesis strategy for novel HPCNs from polymer sources could find use in supercapacitors, lithium ion batteries and fuel cells.
1. Introduction
Over the past several decades, due to the fast worldwide economic expansion and population growth, global energy consumption has been accelerating at an alarming rate.1–3 Consequently, developing not only renewable, clean, and sustainable alternative energies, but also advanced, moderate cost, and eco-friendly energy conversion and storage devices has become an urgent task.4 Supercapacitors have attracted a great deal of attention in energy storage systems because of advantages including a long cycling life, high power density, fast charging–discharge rate, environmental benignity, and good safety.5–8 However, they suffer from low energy density which is less than 10 W h kg−1, an order of magnitude lower than the batteries and fuel cells in practical applications.9 There is an effective approach to improve energy density through increasing the capacitance with high surface area carbon electrode materials and increasing the operation voltage with electrolytes that possess high potential windows.10Recently, hierarchical porous carbon materials can enhance electrochemical charge–discharge process by improving the poor ionic transport of electrolytes in electrode materials.11–15 High surface area microporous carbons enhance the electrical double layer.16 Mesoporous carbons show better charge–discharge rates, especially under high loading current density.17 Besides, the package porosity among carbon nanospheres benefits the generation of ion buffer reservoirs and reduces ion diffusion distance.4,18,19 Since the electrochemical performance is significantly related to the structure of carbon materials, preparation of micro- and mesoporous carbon nanospheres with good dispersibility and well-developed pore structure thus becomes important for achieving good supercapacitive performance.
Activated carbons (ACs) have been the most commonly used electrode materials for electrical double-layer capacitors (EDLCs) over the past years because of their unique properties, while commercially available ACs offer only moderately high specific capacitance.1,12,20 Hydrothermal carbonization is an simple and low cost route for the synthesis of porous materials from natural biomass.21,22 However, biomass-derived ACs usually suffer from significant variation of their properties, which could be limit their application on EDLCs.12 Therefore, there is a critical need to develop a novel methods of ACs synthesis with well-controlled properties and better capacitive characteristics.
Surfactant-free emulsion polymerization is a simple, green process without the addition and subsequent removal of the stabilizing surfactants.23 Herein, we report on the preparation of hierarchical porous carbon nanospheres (HPCNs) from synthetic spherical polymers by surfactant-free emulsion following with one-step KOH activation. The as-prepared ACs materials present favorable features for supercapacitor applications, such as high specific surface area (>3100 m2 g−1), hierarchical porous structure with micro-, meso- and macropores, controlled and tunable pore size distribution and stable thermal and chemical properties. Based on the multiple synergistic effects of these features, HPCNs exhibit superior capacitive performance, including high specific capacitance of 240 F g−1, high cycling life stability (4% loss after 3000 cycles) and high energy density of 77 W h kg−1. In addition, the electrochemical studies are carried out in different electrolytes including 6 M KOH and ethyl-3-methylimidazolium tetrafluoroborate (EMImBF4). It is expected that the studies will be helpful for us to understand the relationship between the pore size of as-prepared materials and capacitive behavior with different electrolytes.
2. Experimental
Preparation of hierarchical porous materials
PAN polymer nanoparticles (PNPs) were prepared by surfactant-free emulsion polymerization of 30 mL acrylonitrile (AN) and 30 mg potassium persulfate in 300 mL distilled water. The reaction was performed at 65 °C for 6 h under protection of N2 with intensive stirring, and then the obtained emulsion was freeze-dried. The dried PNPs powders were subjected to oxidative stabilization by heating in the presence of air to 250 °C for 2 h (named as PNPs-250). To prepare HPCNs sample, the oxidative PNPs powders were mixed with KOH solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 by weight) and stirred for 2 h. The resulting mixture was dried at 120 °C under vacuum for 24 h and then placed in an alumina boat in a tube furnace under N2 flowing and heated to 650–850 °C. The activated carbon samples were washed with 10% acetic acid solution to remove residual inorganic salts and then an excess of distilled water, and the final carbon materials (named as HPCNs-temperature) were created.
:3 by weight) and stirred for 2 h. The resulting mixture was dried at 120 °C under vacuum for 24 h and then placed in an alumina boat in a tube furnace under N2 flowing and heated to 650–850 °C. The activated carbon samples were washed with 10% acetic acid solution to remove residual inorganic salts and then an excess of distilled water, and the final carbon materials (named as HPCNs-temperature) were created.
Fabrication of working electrodes and supercapacitor devices
The working electrodes were prepared by coating the slurry of the active materials (80 wt%), carbon black (10 wt%), and poly(vinylidene fluoride) binder (10 wt%) dispersed in an N-methyl-2-pyrrolidone solvent onto a nickel foam substrate. The weight of active material was calculated from the weight difference between the dried electrode of HPCNs on nickel foam and pure nickel foam of the same size. For ion liquids electrolyte, the slurry was rolled into a uniform film onto Al foil and dried at 80 °C overnight. Then, the electrode was punched into disks with a diameter of 1 cm2. The supercapacitors devices were assembled in a glovebox filled with Ar by constructing two current collectors with active material and a porous film separator sandwiched.Electrochemical measurement of supercapacitor performance
All the electrochemical tests were conducted at room temperature. Cyclic voltammetry (CV), galvanostatic charge/discharge cycle tests, and electrical impedance spectroscope (EIS) studies were performed using CHI760D electrochemical workstation. The gravimetric capacitance (C) was calculated from galvanostatic discharge by using the formula C = (2IΔt)/(mΔV) for the two-electrode cells, where I is the discharge current (A), Δt is the discharge time (s), ΔV is the potential window (V), and m is the mass of active material for single carbon electrodes (g). The energy density (1 W h kg−1 = 3.6 kJ kg−1) was estimated by using the formula E = CV2/8, where V is the cell voltage (V). The power density calculated by using the formula P = E/Δt.Material characterizations
Scanning electron microscopy (SEM) measurement of the samples was investigated using a QUANTA FEG 450 field-emission scanning electron microscopy system. Transmission electron microscopy (TEM) images were recorded on a Hitachi H-8100 TEM system with an accelerating voltage of 200 kV. X-ray diffraction (XRD) patterns of samples were recorded in the 2-theta range from 10 to 80° using a Bruker D8 ADVANCE diffractometer (Germany) with Cu Kα radiation. Infrared spectra were collected on a VERTEX. 70 Fourier transform infrared (FTIR) spectrometer (Bruker). Thermal analysis was conducted using a thermal analyzer (Netzsch STA449 F1) at a heating rate of 10 °C min−1 from 20 to 900 °C in N2. Nitrogen adsorption and desorption isotherms were measured at 77 K and a relative pressure P/P0 of 3.3 × 107 to 0.995 by using a Micromeritics-ASAP-2020. Brunauer–Emmett–Teller (BET) and density functional theory tests were performed to measure the specific surface area and pore size distribution.3. Results and discussion
The preparation of HPCNs is illustrated in Fig. 1 PAN polymer nanospheres were prepared by surfactant-free emulsion polymerization, which is a simple, green process for polymer nanoparticles production without the addition and subsequent removal of the stabilizing surfactants. We selected nitrogen-rich polyacrylonitrile (PAN) as synthetic precursor material due to a high percentage of carbonaceous residue left after thermal treatment. PAN polymer nanoparticles were converted into HPCNs by preoxidation and chemical activation. During the preoxidation treatment, the PAN were chemically stabilized to assure preservation of PAN nanostructure upon chemical activation. The micropores and mesopores were introduced by chemical activation with KOH, while the macropores result from loose aggregation of these nanospheres.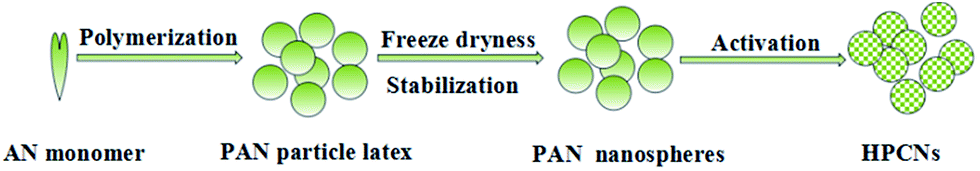 | ||
| Fig. 1 Schematic illustration of the synthesis of HPCNs. | ||
The morphologies of the PAN-derived carbons produced in each step were examined by scanning electron microscopy. SEM images clearly demonstrate that PAN polymer nanospheres exhibited good dispersibility and highly uniform spherical shape about 280 nm in diameter (Fig. 2a). Activated PAN polymer nanospheres (HPCNs-650) preserved the spherical shape with open, interconnected pores in the surface (Fig. 2c and d). The SEM images of HPCNs-750 and HPCNs-850 were given in Fig. S1.† This porous structure is attributed to the preoxidation treatment and KOH activation, which causes PAN polymer nanospheres converted into hierarchical porous carbon nanospheres while preserving the spherical shape of PAN polymer particles. The high-resolution transmission electron microscope (HR-TEM) images (Fig. 2e) further revealed that HPCNs had a highly porous carbon structure with a large fraction of worm-like micro- and small mesopores. Among them, the mesopores can provide a fast diffusion channel and short diffusion distance, and the micropores can enhance the electrical double layer.
 | ||
| Fig. 2 Morphology of as-prepared materials: (a, b and c) SEM images of PNPs, PNPs-250 and HPCNs-650 respectively, (d) TEM image and (e) HR-TEM image of HPCNs-650. | ||
The structure and composition of the materials were investigated by means of FTIR spectroscopy. The data were plotted in Fig. 3a to get information for the structural changes during the preoxidation treatment. FTIR spectrum of PNPs-250 shows a dramatic decrease in the intensity of the 2243 cm−1 (due to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N band) and increase in 1590 cm−1 (due to C
N band) and increase in 1590 cm−1 (due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N band), compared with that of PNPs, which result from intramolecular cyclization or intermolecular crosslinking.24 Moreover, the appearance of the absorption band at 810 cm−1, corresponding to the CC–H stretching vibration, demonstrated the dehydrogenation or imine–enamine tautomerization and subsequent isomerization of PNPs-250 during the stabilization process.25 X-ray diffraction (XRD) has historically played important roles in the structural characterization of carbon materials. The XRD pattern (Fig. 3b) of all samples display broad peaks at 2θ ≈ 25° corresponding to the (002) diffractions, indicating a limited degree of graphitized structure.26 The peaks at 43° represent the (100) diffraction peak, which are consistent with amorphous frameworks. It can be found from Fig. 3b that the intensity of (002) peaks decrease with the increase of temperature, which illustrates that the graphite degree decreases with increasing activation temperature from 650 °C to 850 °C. High-temperature activation create a strong etching action, which is leading to the activation reaction gradually turned on aromatic carbon. Therefore, a large number of amorphous structure with a high specific surface area is formed, which is resulting in a decrease of graphite degree.27 Thermogravimetric analysis of PAN polymer nanoparticles, PNPs-250 and HPCNs-850 in N2 is shown in Fig. 3c. As observed, the approximately 55% and 45% weight losses occur after 850 °C pyrolysis for PNPs and PNPs-250, respectively, which indicated that PNPs-250 has better thermal stability than PNPs.
N band), compared with that of PNPs, which result from intramolecular cyclization or intermolecular crosslinking.24 Moreover, the appearance of the absorption band at 810 cm−1, corresponding to the CC–H stretching vibration, demonstrated the dehydrogenation or imine–enamine tautomerization and subsequent isomerization of PNPs-250 during the stabilization process.25 X-ray diffraction (XRD) has historically played important roles in the structural characterization of carbon materials. The XRD pattern (Fig. 3b) of all samples display broad peaks at 2θ ≈ 25° corresponding to the (002) diffractions, indicating a limited degree of graphitized structure.26 The peaks at 43° represent the (100) diffraction peak, which are consistent with amorphous frameworks. It can be found from Fig. 3b that the intensity of (002) peaks decrease with the increase of temperature, which illustrates that the graphite degree decreases with increasing activation temperature from 650 °C to 850 °C. High-temperature activation create a strong etching action, which is leading to the activation reaction gradually turned on aromatic carbon. Therefore, a large number of amorphous structure with a high specific surface area is formed, which is resulting in a decrease of graphite degree.27 Thermogravimetric analysis of PAN polymer nanoparticles, PNPs-250 and HPCNs-850 in N2 is shown in Fig. 3c. As observed, the approximately 55% and 45% weight losses occur after 850 °C pyrolysis for PNPs and PNPs-250, respectively, which indicated that PNPs-250 has better thermal stability than PNPs.
 | ||
| Fig. 3 Structure of as-prepared materials: (a) FTIR spectra, (b) XRD patterns and (c) thermogravimetric analysis. | ||
The nitrogen absorption–desorption isotherms of the as-prepared materials (Fig. 4a) showed significant impact of the activation temperature on their porosity. The HPCNs-650 sample activated at the lowest temperature of 650 °C exhibits a type-I sorption isotherm with saturation at a relative pressure (P/P0) of ca. 0.1, characteristic for microporous materials with very low volume of pores >2 nm.28 The total amount of N2 adsorbed quantity at P/P0 ≈ 0.99 is ≈482 cm3 g−1, which corresponds to the total pore volume of 0.75 cm3 g−1. As the activation temperature increases to 750 and 850 °C the isotherms change to a type-IV with a pronounced hysteresis in the P/P0 range 0.4–1.0, implying the presence of a large number of small mesopores in HPCNs.29 Moreover, a sharp rise in the N2 isotherm at relatively high pressure from 0.95 to 1.0 (P/P0) indicate the existence of macropores.9,30 As shown in the density functional theory pore size distribution in Fig. 4b, sample HPCNs-650 contained virtually no pores larger than 2 nm. However, activation at higher temperatures of 750 and 850 °C leads to the noticeable increase in the volume of small mesopores in the range of 2–4 nm (see Fig. 4c and d). Such features with hierarchical ordered porous would be beneficial to the ion diffusion and the contact of electrode materials with electrolyte, thus leading to high performance when they were used as the electrode materials for supercapacitors.31–33
 | ||
| Fig. 4 Porosity characterization: (a) nitrogen adsorption–desorption isotherm and (b–d) pore size distribution of polyacrylonitrile-derived activated materials. | ||
Table 1 summarizes the porosity characteristics of our samples. The total BET specific surface area of the carbons was found to increase with activation temperature from 1128 to 2460 and to 3130 m2 g−1. The largest surface areas are higher than the theoretical limit of graphene (2630 m2 g−1).
| Sample | HPCNs-650 | HPCNs-750 | HPCNs-850 |
|---|---|---|---|
| SBET (m2 g−1) | 1128 | 2460 | 3130 |
| Vtotal (cm3 g−1) | 0.75 | 1.57 | 1.87 |
This kind of porous material of activated carbon has several advantages. First, activated carbon nanospheres with micropores and mesopores provide a high surface area, which is benefit to form electric double layers. Second, the uncommon structural characteristics demonstrate multiple advantages for supercapacitor applications, including hierarchical porous to facilitate ion transport, a turbostratic carbon structure to improve electrical conductivity and uniform pore size to optimize charge storage. Third, the thermal and chemical stability of carbon materials can withstand harsh conditions for long periods of time. Fourth, one-step KOH activation is able to decrease the cost of synthesis of the porous carbon materials.
These special features allow HPCNs to serve as a key component in a high-performance electrochemical energy storage supercapacitor. Prior to using the products as the electrode materials for supercapacitors, we first optimized the activation temperatures. We also selected 6 M KOH aqueous electrolyte and EMImBF4 ionic liquids electrolyte to study the capacitive behavior of as-prepared materials.
Fig. 5 shows the electrochemical performance of the supercapacitor with HPCNs as electrodes immersed in 6 M KOH aqueous electrolyte. At scan rate of 50 mV s−1, the cyclic voltammetry curves (Fig. 5a) show quasi-rectangular shape, which is characteristic for an ideal EDLCs with little electrolyte diffusion limitation.34 The galvanostatic charge/discharge curves at 1 A g−1 current density are shown in Fig. 5b. The specific capacitances were calculated on the basis of the discharge curves with values of 240 F g−1, 206 F g−1 and 161 F g−1 for HPCNs-650, HPCNs-750 and HPCNs-850, respectively. Due to the electrical double-layer storage mechanism of carbon materials, high surface areas, more electrolyte ions can be accumulated at the electrode/electrolyte interface.35 Accordingly, the sample with larger surface area obtained higher performance.36 However, the increase in capacitance is relatively limited because not all pores are electrochemically accessible to form the electrical double-layer.1 An adequate pore size is more important than a high surface area to exhibited a large capacitance.37 Generally, narrowing pore size distribution leads to an increase in capacitance.38,39 As observed, HPCNs-650 has a narrower pore size distribution (<2 nm) (Fig. 4b) and larger specific capacitance than HPCNs-750 and HPCNs-850, which contained small mesopores in the range of 2–4 nm. All of the above observation indicate that micropores are electrochemically accessible by KOH electrolyte to form an electrical double-layer and are of great importance to obtain higher specific capacitance. Fig. 5c shows Nyquist plots obtained for all sample in the frequency range of 0.01 Hz to 100 kHz. The Nyquist plots of all sample exhibit the typical features of porous electrodes with a relatively short 45° Warburg region at high frequencies, and an almost vertical line at low frequencies, which the behaviors become mainly capacitive. The projected length of the Warburg-type line (the slope of 45° portion of the curve) on the real axis is related to the diffusion of the ions into the bulk of electrode particles.40 Sample HPCNs-650 showed the largest deviation from the ideal behavior and the largest ionic resistance.
 | ||
| Fig. 5 Electrochemical characterization of PAN-derived ACs materials in KOH electrolyte: (a) CV curves at 50 mV s−1, (b) galvanostatic charge/discharge cures at 1 A g−1 and (c) Nyquist plots, the inset shows a magnified view of the high-frequency region. | ||
Fig. 6 shows the electrochemical performance of the supercapacitor with HPCNs as electrodes immersed in ionic liquid of EMImBF4 electrolyte. The cyclic voltammetry curves (Fig. 6a) also show quasi-rectangular shape at scan rate of 50 mV s−1. Fig. 6b presents the galvanostatic charge/discharge curves in the voltage range of 0–3.5 V with specific capacitances of 86, 126, and 181 F g−1 at current density of 1 A g−1, which is corresponded to the product of HPCNs-650, HPCNs-750 and HPCNs-850, respectively. It can be found that the specific capacitances increased with increased carbonization temperatures, which is different from the results of KOH as electrolyte. This difference may be related to the porosity of carbon materials and the molecular size of the electrolyte, which is the easier accessibility of the larger size of charged electrolyte molecules to large micropores of carbon materials.
 | ||
| Fig. 6 Electrochemical characterization of PAN-derived ACs materials in EMImBF4 electrolyte: (a) CV curves at 50 mV s−1, (b) galvanostatic charge/discharge curves at 1 A g−1. | ||
We proceeded to investigate the electrochemical properties in EMImBF4 electrolyte of the most promising carbon electrode sample, HPCNs-850, in details. The CV curves were measured with various scan rates ranging from 50 to 200 mV s−1 (Fig. 7a). The almost rectangular CV curves at fast sweep rate of 200 mV s−1 indicated very efficient charge transfer within the porous electrodes.41 Furthermore, the galvanostatic charge/discharge curves at different current densities from 2 A g−1 to 10 A g−1 were all similar to a linear shape within the potential window between 0 V and 3.5 V (see Fig. 7b). Fig. 7c summarizes the effect of the different current densities (from 1 A g−1 to 10 A g−1) in galvanostatic charge–discharge tests on the specific capacitance. The experimentally observed decrease of capacitance at higher current, which is generally attributed to the resistance of ions traveling within the nanopores.12 In order to confirm the cycle stability of the as-prepared HPCNs-850, galvanostatic charge–discharge tests at the constant current density of 5 A g−1 up to 3000 cycles were carried out (see Fig. 7d). Remarkably, the as-prepared carbon materials hold the high specific capacitance retention of 96% even after 3000 cycles. The SEM image of HPCNs-850 after 3000 cycles was given in Fig. S2.† The Ragone plots for symmetrical supercapacitors displayed in Fig. 7e clearly demonstrate the high energy density and power performance. Benefitting from a specific capacitance up to 181 F g−1 at a current density of 1 A g−1 and an operating voltage of 3.5 V, the device exhibited the high gravimetric energy density of 77 W h kg−1 and a gravimetric power density of 875 W kg−1. Table 2 shows a comparison of the supercapacitor performance of typical hierarchical porous carbon materials reported in the literature. As observed, the as-prepared materials have some advantages in supercapacitor performance.
 | ||
| Fig. 7 Electrochemical characterization of HPCNs-850 in EMImBF4 electrolyte: (a) CV curves at various scan rates, (b) galvanostatic charge/discharge curves at different current densities, (c) capacitance retention with current densities from 1 A g−1 to 10 A g−1, (d) cycling stability at a current density of 5 A g−1, and (e) Ragone plots showing energy density and power density. | ||
| Precursor | SBET (m2 g−1) | Vtotal (cm3 g−1) | Electrolyte | Chigh (F g−1) | Energy density (W h kg−1) | Cell | Ref. |
|---|---|---|---|---|---|---|---|
| UCNs | 842 | 0.74 | KOH | 206 (1 A g−1) | — | 3E | 42 |
| CH4 | 140 | 0.337 | KOH | 20 (0.2 A g−1) | — | 3E | 43 |
| POF | 525 | 0.67 | KOH | 230 (0.5 A g−1) | — | 3E | 44 |
| Furfuryl alcohol | 709 | 0.88 | KOH | 221 (1 A g−1) | — | 3E | 45 |
| Phenolic | 545 | 0.26 | KOH | 256 (0.2 A g−1) | — | 2E | 46 |
| Melanine | 8 | — | KOH | 198 (0.1 A g−1) | — | 2E | 47 |
| RGO-3 | 2406 | 1.97 | EMImBF4 | 131.5 (1 mV s−1) | 57.5 | 2E | 48 |
| BHNC | 1472 | 0.61 | EMImTFSI | 146 (0.2 A g−1) | 43.3 | 2E | 49 |
| HPCNs-650 | 1128 | 0.75 | KOH | 240 (1 A g−1) | — | 2E | — |
| HPCNs-850 | 3130 | 1.87 | EMImBF4 | 181 (1 A g−1) | 77 | 2E | — |
4. Conclusions
In summary, we have successfully developed a green, simple, template-free approach to fabricate hierarchically porous carbon nanospheres from synthetic precursors. The as-obtained HPCNs possess hierarchical micro-, meso-, macroporous pore structure, and exhibits a large BET surface area in the range of 1128–3130 m2 g−1 and pore volume in the range of 0.75–1.87 cm3 g−1. Activation at high temperatures lead to increase in the surface area of the carbons and the broadening of the pore size distribution through the formation of additional volume of 1–4 nm pores. These unique properties have favourable multiple synergistic effects, beneficial for excellent supercapacitor performance. For a two-electrode system, the HPCNs shows a specific capacitance of 240 F g−1, and good stability of 96% capacitance retention after 3000 cycles. Moreover, the assembled symmetric supercapacitor displays a high energy density of 77 W h kg−1 at a high power density of 875 W kg−1. We also concluded that the pore sizes of carbon materials match with those of the electrolyte ion sizes could result in the maximum specific capacitance, which suggests a general approach to selecting a porous electrode/electrolyte couple. This hierarchical structure and its high specific surface area could find use in supercapacitors with high power and energy density.Acknowledgements
We gratefully acknowledge the support of the National Science Foundation of China (51102169, 51272157, 51102168) and the Hujiang Foundation of China (B14006).Notes and references
- J. Yan, Q. Wang, T. Wei and Z. Fan, Adv. Energy Mater., 2014, 4, 1300816 Search PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- M. Liu, L. Gan, W. Xiong, Z. Xu, D. Zhu and L. Chen, J. Mater. Chem. A, 2014, 2, 2555–2562 CAS.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 28, 2520–2531 RSC.
- C. Xu, J. Sun and L. Gao, J. Mater. Chem., 2011, 21, 11253–11258 RSC.
- Y. Li, C. Lu, S. Zhang, F. Y. Su, W. Shen, P. Zhou and C. Ma, J. Mater. Chem. A, 2015, 3, 14817–14825 CAS.
- B. Chang, Y. Guo, Y. Li, H. Yin, S. Zhang, B. Yang and X. Dong, J. Mater. Chem. A, 2015, 3, 9565–9577 CAS.
- Q. Zhao, X. Wang, J. Liu, H. Wang, Y. Zhang and J. Gao, Electrochim. Acta, 2015, 154, 110–118 CrossRef CAS.
- P. Hao, J. Tian, H. Li, Y. Sang, G. Yu, H. Cai, C. P. Wong and A. Umar, Nanoscale, 2014, 6, 12120–12129 RSC.
- J. Zhang, L. Jin, J. Cheng and H. Hu, Carbon, 2013, 55, 221–232 CrossRef CAS.
- L. Wei, M. Sevilla, A. B. Fuertes, R. Mokaya and G. Yushin, Adv. Funct. Mater., 2012, 22, 827–834 CrossRef CAS.
- Q. Zhao, X. Wang, H. Xia, J. Liu, H. Wang, J. Gao, Y. Zhang, J. Liu, H. Zhou, X. Li, S. Zhang and X. Wang, Electrochim. Acta, 2015, 173, 566–574 CrossRef CAS.
- C. Wu, J. Gao, Q. Zhao, Y. Zhang, Y. Bai, X. Wang and X. Wang, J. Power Sources, 2014, 269, 818–824 CrossRef CAS.
- J. Gao, X. Wang, Q. Zhao, Y. Zhang and J. Liu, Electrochim. Acta, 2015, 163, 223–231 CrossRef CAS.
- J. Chmiola, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- W. Xing, S. Z. Qiao, R. G. Ding, F. Li, G. Q. Lu and Z. F. Yan, Carbon, 2006, 44, 216–224 CrossRef CAS.
- Y. Lv, L. Gan, M. Liu, W. Xiong, Z. Xu, D. Zhu and D. S. Wright, J. Power Sources, 2012, 209, 152–157 CrossRef CAS.
- F. Xu, R. Cai, Q. Zeng, C. Zou, D. Wu and F. Li, J. Mater. Chem., 2011, 21, 1970–1976 RSC.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- M. M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC.
- M. M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36887–36913 Search PubMed.
- S. Dalton, F. Heatley and P. M. Budd, Polymer, 1999, 40, 5531–5543 CrossRef CAS.
- W. Zhang, J. Liu and G. Wu, Carbon, 2003, 41, 2805–2812 CrossRef CAS.
- C. Zou, D. Wu, M. Li, Q. Zeng, F. Xu and Z. Huang, J. Mater. Chem., 2010, 20, 731–735 RSC.
- X. He, J. Lei, Y. Geng, X. Zhang, M. Wu and M. Zheng, J. Phys. Chem. Solids, 2009, 70, 738–744 CrossRef CAS.
- T. Y. Kim, G. Jung, S. Yoo, K. S. Suh and R. S. Ruoff, ACS Nano, 2013, 7, 6899–6905 CrossRef CAS PubMed.
- H. Feng, M. Zheng, H. Dong, Y. Xiao, H. Hu, Z. Sun, C. Long, Y. Cai, X. Zhao, H. Zhang, B. Lei and Y. Liu, J. Mater. Chem. A, 2015, 3, 15225–15234 CAS.
- K. T. Lee, J. C. Lytle, N. S. Ergang, S. M. Oh and A. Stein, Adv. Funct. Mater., 2005, 15, 547–556 CrossRef CAS.
- B. G. Choi, M. H. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
- S. Dutta, A. Bhaumik and K. C. W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 CAS.
- X. Cao, Y. Shi, W. Shi, G. Lu, X. Huang, Q. Yan, Q. Zhang and H. Zhang, Small, 2011, 7, 3163–3168 CrossRef CAS PubMed.
- F. Wu, L. Shi, D. Mu, H. Xu and B. Wu, Carbon, 2015, 86, 146–155 CrossRef CAS.
- K. Xie, X. Qin, X. Wang, Y. Wang, H. Tao, Q. Wu, L. Yang and Z. Hu, Adv. Mater., 2012, 24, 347–352 CrossRef CAS PubMed.
- Y. M. Chang, C. Y. Wu and P. W. Wu, J. Power Sources, 2013, 223, 147–154 CrossRef CAS.
- E. R. Pinero, K. Kierzek, J. Machnikowski and F. Beguin, Carbon, 2006, 44, 2498–2507 CrossRef.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- C. Largeot, C. Portet, J. Chmiola, P. L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- X. Yang, J. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833–2838 CrossRef CAS PubMed.
- L. L. Zhang, X. Zhao, M. D. Stoller, Y. Zhu, H. Ji, S. Murali, Y. Wu, S. Perales, B. Clevenger and R. S. Ruoff, Nano Lett., 2012, 12, 1806–1812 CrossRef CAS PubMed.
- Y. Zhao, M. Liu, L. Gan, X. Ma, D. Zhu, Z. Xu and L. Chen, Energy Fuels, 2014, 28, 1561–1568 CrossRef CAS.
- A. Śliwak, B. Grzyb, J. Cwikła and G. Gryglewicz, Carbon, 2013, 64, 324–333 CrossRef.
- X. Liu, L. Zhou, Y. Zhao, L. Bian, X. Feng and Q. Pu, ACS Appl. Mater. Interfaces, 2013, 5, 10280–10287 CAS.
- Q. Zhao, X. Wang, C. Wu, J. Liu, H. Wang, J. Gao, Y. Zhang and H. Shu, J. Power Sources, 2014, 254, 10–17 CrossRef CAS.
- C. Ma, Y. Song, J. Shi, D. Zhang, X. Zhai, M. Zhong, Q. Guoand and L. Liu, Carbon, 2013, 51, 290–300 CrossRef CAS.
- D. Hulicova-Jurcakova, M. Kodama, S. Shiraishi, H. Hatori, Z. H. Zhu and G. Q. Lu, Adv. Funct. Mater., 2009, 19, 1800–1809 CrossRef CAS.
- C. zheng, X. F. Zhou, H. L. Gao, G. H. Wang and Z. P. Liu, J. Mater. Chem. A, 2015, 3, 9543–9549 CAS.
- W. Tian, Q. Gao, Y. Tan, K. Yang, L. Zhu, C. Yang and H. Zhang, J. Mater. Chem. A, 2015, 3, 5656–5664 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2. See DOI: 10.1039/c5ra27000f |
| This journal is © The Royal Society of Chemistry 2016 |