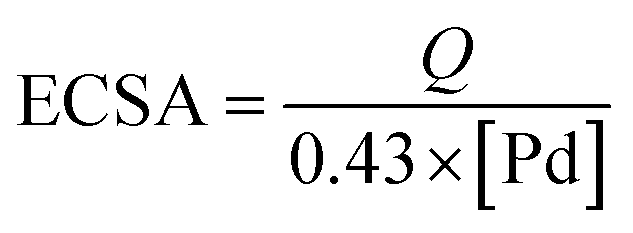DOI:
10.1039/C5RA26994F
(Paper)
RSC Adv., 2016,
6, 19314-19321
One-step synthesis of nitrogen-doped graphene supported PdSn bimetallic catalysts for ethanol oxidation in alkaline media
Received
17th December 2015
, Accepted 10th February 2016
First published on 12th February 2016
Abstract
In this paper, a facile chemical reduction method was employed to synthesize bimetallic PdSn nanocatalysts, and the nitrogen-doped graphene (N-G) has been used as a conductive support material for PdSn catalysts. The structures, morphologies and composition of the catalysts were characterized by transmission electron microscopy (TEM), energy dispersive X-ray spectroscopy (EDX), X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS), respectively. Electrochemical measurements such as cyclic voltammetry (CV), chronoamperometry (CA), electrochemical impedance spectrometry (EIS) were carried out to detect the electrochemical activity of the as-prepared catalysts toward ethanol electrooxidation in an alkaline medium. Among the PdSn/N-G catalysts with different molar ratios between Pd and Sn, the free-standing PdSn catalyst and commercial Pd/C catalyst, Pd3Sn1/N-G exhibited excellent catalytic activity and stability, indicating that nitrogen-doped graphene as a conductive support material and the moderate addition of Sn can enhance the electrochemical performance of Pd catalysts.
Introduction
Direct ethanol fuel cells (DEFCs) are well-known promising energy converters that have attracted widespread attention for electric vehicles and portable electronic devices due to their high theoretical energy density, low operating temperature, low toxicity, abundant availability and environmental friendliness.1 However, complete electrooxidation of ethanol to CO2 poses a practical challenge for anode catalysts.2 Pd is one of the most active materials for the ethanol oxidation reaction since it is sufficiently reactive at low temperature; however, Pd is expensive and non-abundant as a noble metal, which has been a bottleneck for large-scale commercial application for the electrochemical oxidation of ethanol.3 On the one hand, it is necessary to find catalysts with minimum Pd loading, for example, introducing new components, such as ruthenium (Ru), cobalt (Co), nickel (Ni) and stannum (Sn) into Pd catalysts to utilize the binary metal effect to achieve a good tolerance of CO poisoning.4,5 Among Pd based bimetallic catalysts, the PdSn system displays better electrochemical activity because Sn atoms can promote the oxidation of CO on Pd via bimetallic synergistic effect.6 What's more, Sn is inexpensive compared with the noble metal Pd.7 On the other hand, developing appropriate supporting materials is also a good way to provide the Pd catalysts with greater catalytic activity and stability.8,9 As known, various kinds of carbon materials have been used as supporting materials for fuel cell catalysts, such as carbon nanotubes (CNT), carbon fiber (CF), Vulcan XC-72 carbon black, graphitized carbon materials and graphene.10–12 Particularly, graphene exhibits many unique physical and chemical properties, including its high specific surface area, good electronic conductivity, chemical stability and ultrathin thickness, which offers attractive benefits as an advanced supporting material for electrochemical oxidation catalysts.13–15 Pd catalysts supported on graphene have been studied with great performance for methanol/ethanol oxidation reaction.16 To further enhance the properties of graphene, nitrogen doping method has been widely applied.17 The heteroatoms of nitrogen can induce a charge rearrangement on the graphene sheets, consequently create new catalytic centres.17 Nitrogen atoms provide the enormous impact on binding force between nanoparticles and grapheme, and the surface charge distribution is changed and more anchoring sites are created for nanoparticles.18,19 Moreover, the incorporation heterogeneous nitrogen atoms into the graphene which can reduces the particle size and improves the dispersion. Thus, nitrogen-doped graphene (N-G) act as a better support materials than graphene for anchoring catalysts.20 Thus, nitrogen-doped graphene (N-G) act as a better support materials than graphene for anchoring catalysts. For instance, Dong et al. have indentified that the catalytic activity of Pt NPs loaded on N-G was much higher than that loaded on undoped graphene for oxygen reaction and methanol oxidation.21
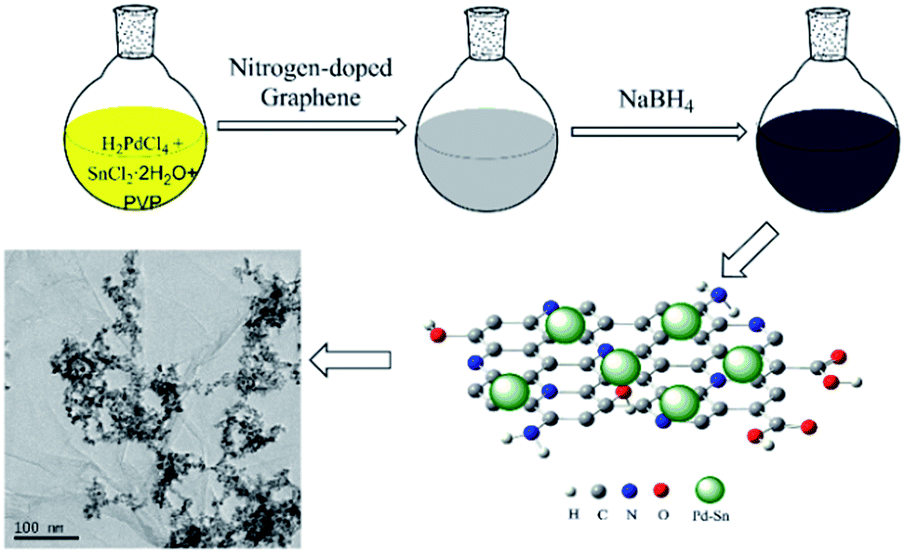
In this work, we report a facile synthesis of PdSn bimetallic catalysts for ethanol oxidation reaction, using nitrogen-doped graphene as a supporting material (as shown in Scheme 1). Firstly, nitrogen-doped graphene (N-G) was dispersed in dimethylformamide (DMF) and stirred for 30 min to produce a homogeneous solution. Then H2PdCl4 solution and SnCl2 solution were mixed together, and polyvinylpyrrolidone (PVP) were dissolved in the metal precursor aqueous solution by vigorous stirring to increase the dissolution of PVP. Subsequently, the N-doped graphene suspension was dropwise added to the mixture. Freshly prepared NaBH4 solution was used as reducing agent at the room temperature. PdSn composite catalysts dispersed onto the N-G supports are studied to examine their electrochemical catalytic activity for ethanol electrooxidation in an alkaline medium compared with other free-standing PdSn catalysts and commercial Pd/C catalyst, and the optimum molar ratio for the PdSn composite catalyst is also studied in this work. The results indicate that the amount addition of Sn can obviously affect the catalytic activity of Pd catalysts, and the N-G supports can also enhance the activity of the as-prepared catalysts.
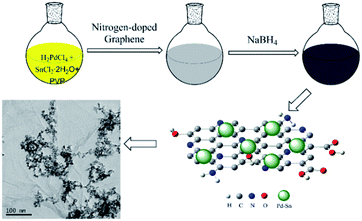 |
| | Scheme 1 Illustration of the synthesis for PdSn/N-G catalysts. | |
Experimental section
Materials and instruments
Nitrogen-doped graphene (Nanjing XFNANO Materials Tech Co., Ltd), DMF (Shanghai Shiyi Chemicals Regent Co., Ltd, China), H2PdCl4 (Shanghai Shiyi Chemicals Regent Co., Ltd, China), SnCl2·2H2O (Shanghai Shiyi Chemicals Reagent Co., Ltd, China), polyvinylpyrrolidone (PVP, K35), NaBH4, C2H5OH and KOH (Sinopharm Chemical Reagent Co., Ltd, China) were all of analytical grade and used without further purification. Commercial Pd/C catalyst (JM 20% Pd) were purchased from Shanghai Hesen Electric Co., Ltd, China. Doubly distilled water was used throughout the experiments.
All the electrochemical experiments were carried out on a CHI 760E potentiostat/galvanostat (CH Instrumental Co., Ltd, China) with a conventional three-electrode-cell system at room temperature. A glassy carbon electrode (GCE, 3 mm diameter) modified with as-prepared catalysts solution was used as working electrode. A Pt wire was used as the counter electrode and the saturated calomel electrode (SCE) was used as the reference electrode. Cyclic voltammetry (CV) measurement was used to analyze the catalytic activity of as-prepared catalysts in an alkaline media containing 1.0 M KOH and 1.0 M C2H5OH solution with a scan rate of 50 mV s−1. Chronoamperometry (CA) measurements were conducted at −0.30 V in a solution of 1.0 M KOH and 1.0 M C2H5OH for 3600 s. The electrochemical impedance spectrometry (EIS) was carried out between 1 Hz and 105 Hz with the AC voltage amplitude 5.0 mV.
Synthesis of the as-prepared catalysts
The nitrogen-doped graphene PdSn (PdSn/N-G) catalysts were synthesized by a simple reduction method. The preparation process follows: 10 mg nitrogen-doped graphene was dissolved in 20 mL DMF, and the mixture was stirred for 30 min. The metal precursor aqueous solution containing 0.5 mL H2PdCl4 (22.56 mM) solution and amount of SnCl2·2H2O (8.8 mM) solution were mixed together under the constant stirring. The 60 mg PVP were dissolved in the metal precursor aqueous solution by vigorous stirring. N-G (0.5 mg mL−1) solution 1 mL was added into the mixture. Subsequently, 2 mL freshly prepared NaBH4 (8 mg mL−1) aqueous solution was added dropwise into the prepared solution under constant stirring for 3 h at room temperature. The black suspension was centrifuged and washed several times with doubly distilled water and alcohol. Then, the black powder was dispersed in 5 mL doubly distilled water. After ultrasonic homogenization of the suspension, catalyst “ink” was obtained. The as-prepared catalysts were denoted as Pd0.5Sn1/N-G, Pd1Sn1/N-G, Pd2Sn1/N-G, Pd3Sn1/N-G, Pd4Sn1/N-G. For comparison, the Pd/N-G and Pd3Sn1 catalyst was synthesized by the same procedure. All the catalysts contain a total Pd loading 2.4 mg.
Characterization
X-ray diffraction (XRD) analysis was carried out on a PANalytical X'Pert PRO MRD X-ray diffractometer (PANalytical Company, the Netherlands) with Cu Kα radiation (λ = 1.54056 Å) operated at 40 kV and 30 mA. An energy-dispersive X-ray analyzer (EDX, S-4700, Japan) and a TecnaiG220 transmission electron microscope (TEM) were used to characterized composition and structure of the samples. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific ESCALab 250Xi using 200 W monochromated Al Kα radiation.
Results and discussion
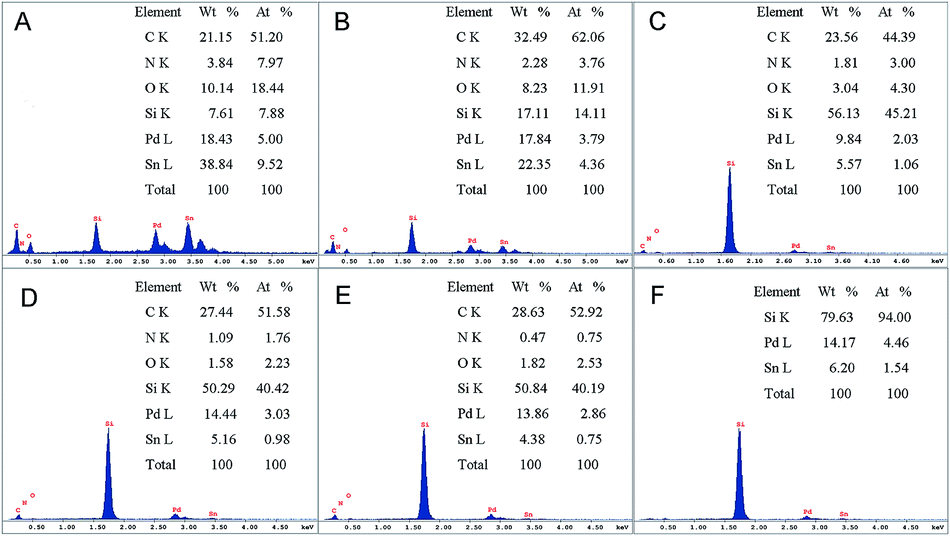
The EDX spectrum presented in Fig. 1 were performed to analyse the surface composition of all the as-prepared catalysts. The molar ratio of Pd to Sn in catalysts Pd0.5Sn1/N-G, Pd1Sn1/N-G, Pd2Sn1/N-G, Pd3Sn1/N-G, Pd4Sn1/N-G and Pd3Sn1 is measured to be 0.5, 0.9, 1.9, 3.1, 3.7 and 2.9, respectively, which is nearly consistent with the molar ratio of Pd and Sn precursors before reduction. The results reveal that all the catalysts with different molar ratio between Pd and Sn have been obtained, which means that a simple chemical reduction method is reliable and suitable in this work. Moreover, the peaks of C, N, O in the spectra indicate the existence of nitrogen-doped graphene.
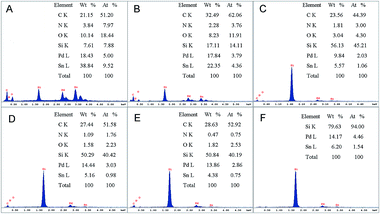 |
| | Fig. 1 EDX spectra for Pd0.5Sn1/N-G (A), Pd1Sn1/N-G (B), Pd2Sn1/N-G (C), Pd3Sn1/N-G (D), Pd4Sn1/N-G (E) and Pd3Sn1 (F). | |
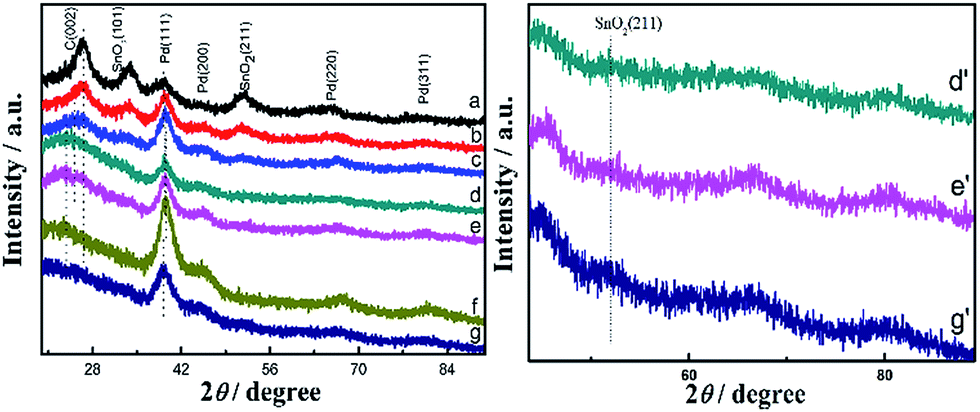
The crystal structures of the as-prepared catalysts were confirmed using X-ray diffraction (XRD) technique. The peaks at around 26.6°, 33.8° and 51.8° can be ascribed to the (110), (101) and (211) faces of the SnO2 structure, respectively.4,5,22 The peak at around 23.8° is associated with the (002) plane of carbon in Fig. 2.23 It can be observed that the C (002) planes of nitrogen-doped graphene in the as-prepared catalysts were consistently shifted to higher 2θ value as the amount of Sn increased, from curve e to curve a in Fig. 2. SnO2 (110) peak at about 26.5° partially overlaps with C (002) peak at around 23.8°, resulting in the higher intensity and higher 2θ value of C (002) peak in the Pd0.5Sn1/N-G catalyst than other catalysts. What's more, the intensity of SnO2 peaks becomes weak when the amount of Sn is reduced. The peaks at around 39.7°, 45.8°, 67.7° and 81.3° are attributed to the diffraction peaks of face-centered cubic (fcc) planes of Pd (111), Pd (200), Pd (220) and Pd (311).24,25 However, the 2θ values of the (111) peak for Pd0.5Sn1/N-G, Pd1Sn1/N-G, Pd2Sn1/N-G, Pd3Sn1/N-G, Pd4Sn1/N-G and Pd3Sn1 catalysts were slightly shifted, compared with Pd/N-G catalyst, suggesting that the Sn has been introduced into the Pd forming PdSn alloy.26 However, Sn is easily oxidized to tin oxide in store, so the diffraction peaks of SnO2 are observed in the image. The diffraction peaks of Sn are rarely observed in the composites, which may be attributed to their relatively low content of Sn in the catalysts or the replacement of lattice positions.
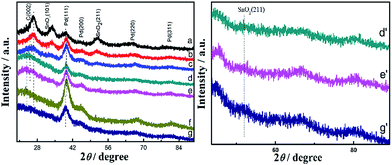 |
| | Fig. 2 XRD patterns of Pd0.5Sn1/N-G (a), Pd1Sn1/N-G (b), Pd2Sn1/N-G (c), Pd3Sn1/N-G (d), Pd4Sn1/N-G (e), Pd/N-G (f) and Pd3Sn1 (g). Enlarged patterns of d′ (Pd3Sn1/N-G), e′ (Pd4Sn1/N-G) and g′ (Pd3Sn1). | |
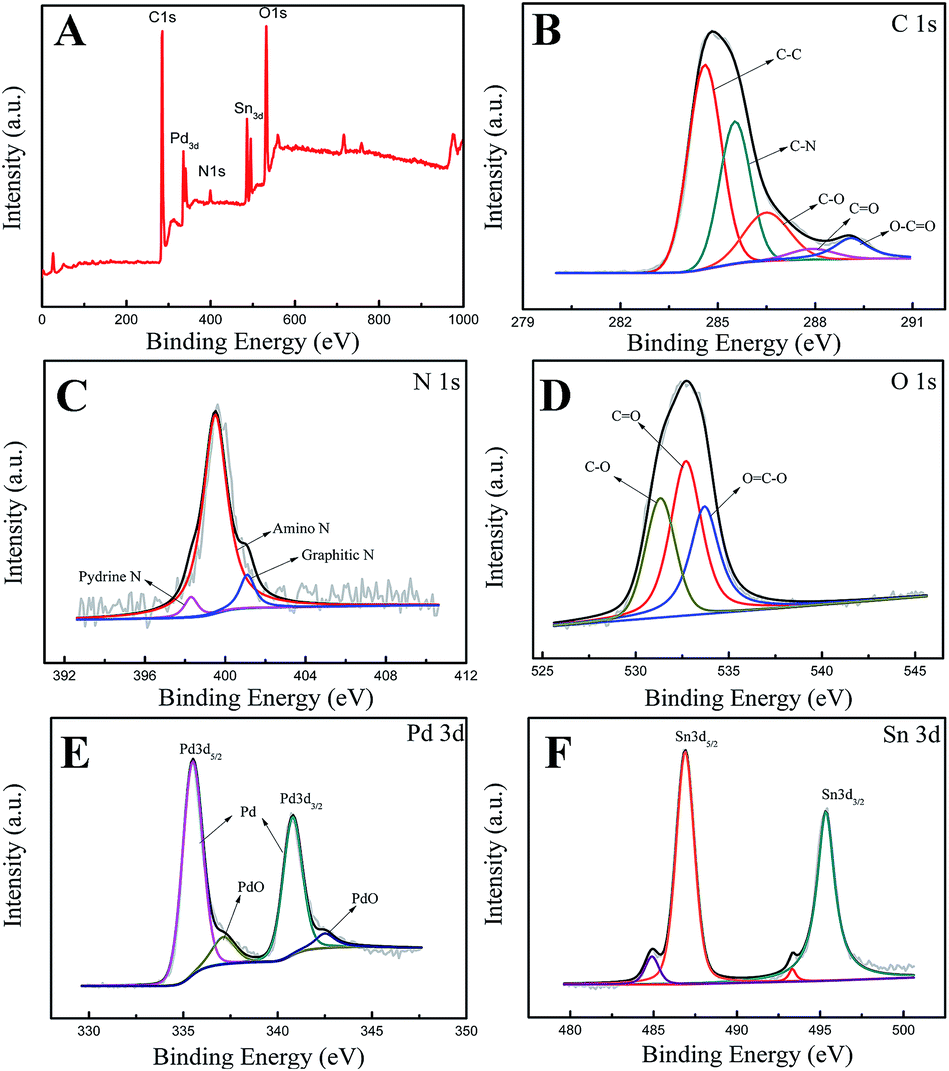
Fig. 3 shows the XPS spectra of the Pd3Sn1/N-G catalysts, and the elemental compositions of the as-prepared catalyst were presented in Table 1. The C 1s spectrum of Pd3Sn1/N-G in Fig. 3B could be reasonably fitted with five components corresponding to the following carbon functional groups: C–C (284.5 eV), C–N (285.5 eV), C–O (286.5 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (287.3 eV) and O–CO (289.1 eV).27,28 Fig. 3C shows the N 1s spectrum of the catalysts deconvoluted with three types of nitrogen: pyridinic-type N (398.3 eV), amino-type N (399.5 eV) and graphitic-type N (401.1 eV).29,30 The spectrum of O 1s displayed in Fig. 3D could be reasonably deconvoluted into three peaks: C–O, CO and OC–O at the respective binding energy of 531.3 eV, 532.7 eV and 533.8 eV.30 Apparently, the two distinct peaks located at 335.6 eV and 340.8 eV can be assigned to the Pd 3d5/2 (high-energy band) and Pd 3d3/2 (low-energy band) spin orbit states of zero-valent Pd, and the less intense doublets around 337.1 eV and 342.5 eV can be ascribed to 3d3/2 and 3d5/2 peaks of PdO.9,23,31 Fig. 3F exhibits the Sn 3d region of XPS spectra of Pd3Sn1/N-G catalyst. The result indicates that two states of Sn exist in the catalyst, one is Sn alloy Sn0, the other is SnOx. The Sn3d spectra clearly exhibited intense doublets attributable to the 3d3/2 (495.3 eV) and 3d5/2 (486.9 eV) of Sn2+/4+.6,32 In addition, it is difficult to distinguish between Sn2+ and Sn4+ by XPS, since their binding energies are very close.33 According to the characterization of XRD, Sn has been oxidized to SnO2. Other researchers also found that most tin atoms existed as oxides in Pd/Sn catalysts prepared by different methods. The metallic Sn was also observed in the image, 3d5/2 (485 eV) and 3d3/2 (493.4 eV).5,34 However, the lower content of metallic Sn can be observed in Fig. 3F, which can explain why no obvious metallic Sn peaks in XRD patterns. Combined with XRD and XPS, we conclude that the catalyst is PdSn alloy, while Sn atoms on the surface are partially oxided into SnO2.
O (287.3 eV) and O–CO (289.1 eV).27,28 Fig. 3C shows the N 1s spectrum of the catalysts deconvoluted with three types of nitrogen: pyridinic-type N (398.3 eV), amino-type N (399.5 eV) and graphitic-type N (401.1 eV).29,30 The spectrum of O 1s displayed in Fig. 3D could be reasonably deconvoluted into three peaks: C–O, CO and OC–O at the respective binding energy of 531.3 eV, 532.7 eV and 533.8 eV.30 Apparently, the two distinct peaks located at 335.6 eV and 340.8 eV can be assigned to the Pd 3d5/2 (high-energy band) and Pd 3d3/2 (low-energy band) spin orbit states of zero-valent Pd, and the less intense doublets around 337.1 eV and 342.5 eV can be ascribed to 3d3/2 and 3d5/2 peaks of PdO.9,23,31 Fig. 3F exhibits the Sn 3d region of XPS spectra of Pd3Sn1/N-G catalyst. The result indicates that two states of Sn exist in the catalyst, one is Sn alloy Sn0, the other is SnOx. The Sn3d spectra clearly exhibited intense doublets attributable to the 3d3/2 (495.3 eV) and 3d5/2 (486.9 eV) of Sn2+/4+.6,32 In addition, it is difficult to distinguish between Sn2+ and Sn4+ by XPS, since their binding energies are very close.33 According to the characterization of XRD, Sn has been oxidized to SnO2. Other researchers also found that most tin atoms existed as oxides in Pd/Sn catalysts prepared by different methods. The metallic Sn was also observed in the image, 3d5/2 (485 eV) and 3d3/2 (493.4 eV).5,34 However, the lower content of metallic Sn can be observed in Fig. 3F, which can explain why no obvious metallic Sn peaks in XRD patterns. Combined with XRD and XPS, we conclude that the catalyst is PdSn alloy, while Sn atoms on the surface are partially oxided into SnO2.
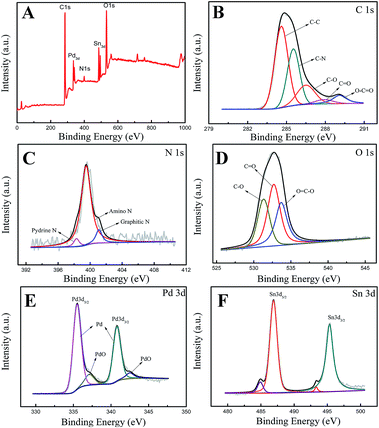 |
| | Fig. 3 XPS spectra of the Pd3Sn1/N-G catalyst (A); XPS core level spectra of the C 1s (B), N 1s (C), O 1s (D), Pd 3d (E) and Sn 3d (F). | |
Table 1 Fitting results of C 1s, N 1s, O 1s, Pd 3d and Sn 3d core level XPS spectra of Pd3Sn1/N-G catalyst
| Sample |
Carbon functional group (BE, eV) |
Nitrogen functional group (BE, eV) |
|
| Pd3Sn1/N-G |
C–C |
C–N |
C–O |
CO |
O–CO |
Pyridine N |
Amino N |
Graphitic N |
|
| 284.5 |
285.5 |
286.5 |
287.3 |
289.1 |
398.3 |
399.5 |
401.1 |
|
| Sample |
Oxygen functional group (BE, eV) |
| Pd3Sn1/N-G |
C–O |
CO |
OC–O |
| 531.3 |
532.7 |
533.8 |
| Sample |
Pd species (BE, eV) |
Sn species (BE, eV) |
| Pd3Sn1/N-G |
Pd03d5/2 |
Pd2+3d5/2 |
Pd03d3/2 |
Pd2+3d3/2 |
Sn4+3d5/2 |
Sn03d5/2 |
Sn4+3d3/2 |
Sn03d3/2 |
| 335.6 |
337.1 |
340.8 |
342.5 |
485 |
486.9 |
493.4 |
495.3 |
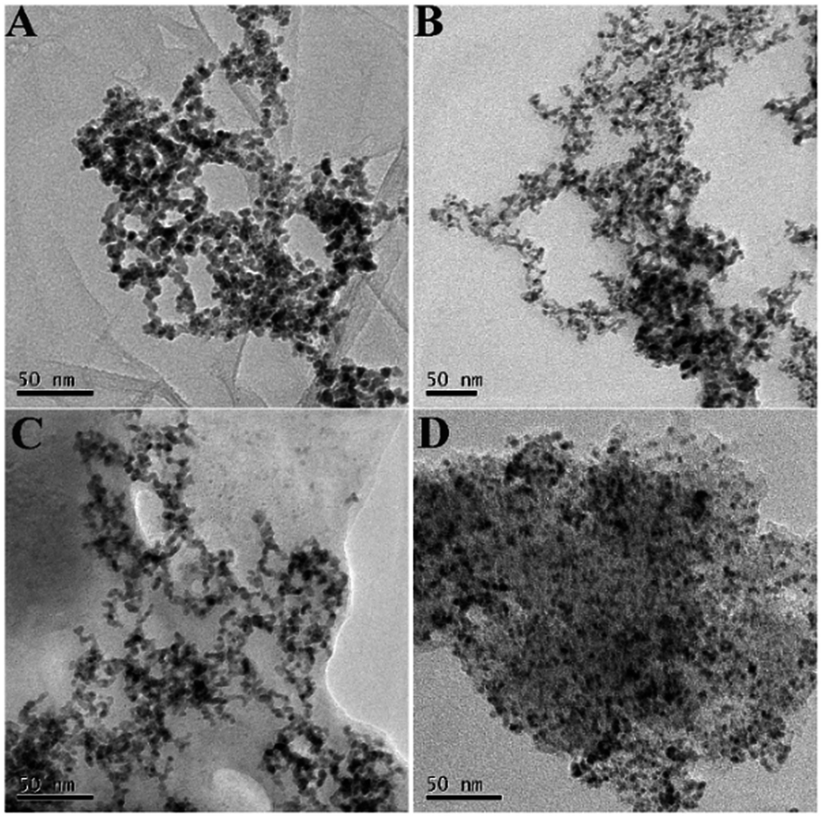
The network structures of Pd3Sn1/N-G (A), Pd3Sn1 (B) and Pd/N-G (C) were characterized by transmission electron microscopy (TEM) shown in Fig. 4. All the as-prepared catalysts presented the same network structure, and the nanoparticles are fused to compose the network. The result indicates that the composition of Sn and the supporting of N-G will not change the morphology of the network structure of the catalysts. As known, a primary purpose of the introducing of PVP is to protect the metal nanoparticles from growing and agglomerating. With the introduction of PVP, PdSn particles would coordinate with N or O in PVP and a covered layer would generate on the surface of the particles. The layer inhibited the growth and agglomeration of the particles.35 The PVP contained lots of positive charges due to the protonated carbonyl groups, therefore electrostatic repulsion led to the formation of dispersed PdSn nanoparticles. The PVP polymer chains connected to each other easily, so the PdSn nanochains formed networks.36 Moreover, the addition of N-G increases the dispersion of metal catalysts, which can enhance the catalytic activity of catalysts for ethanol oxidation reaction. In the commercial Pd/C composite, a large number of nanoparticles agglomerated together shown in Fig. 4D, which could have a bad effect on the electrochemical catalytic activity of commercial Pd/C catalysts.
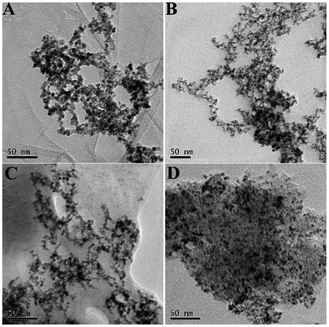 |
| | Fig. 4 TEM images of Pd3Sn1/N-G (A), Pd3Sn1 (B), Pd/N-G (C) and commercial Pd/C (D). | |
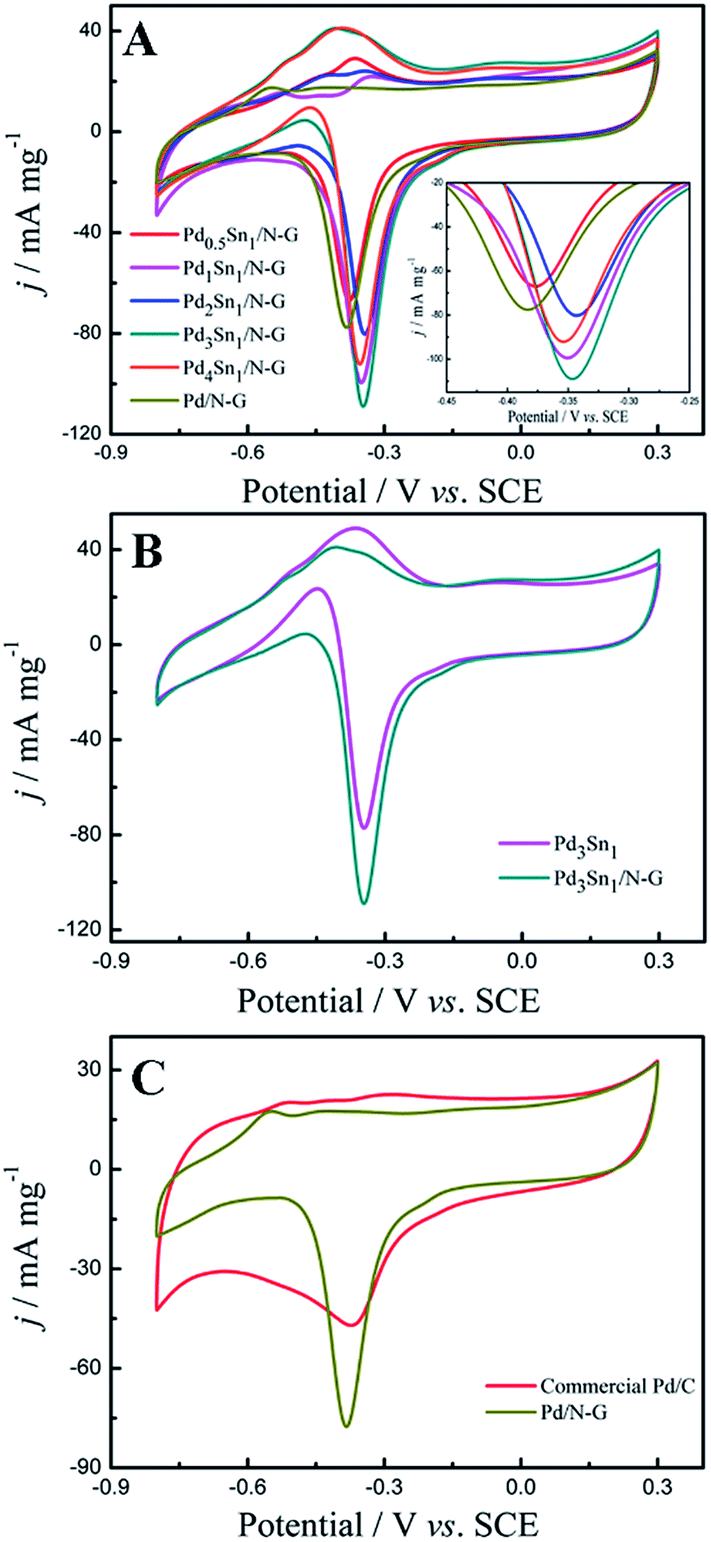
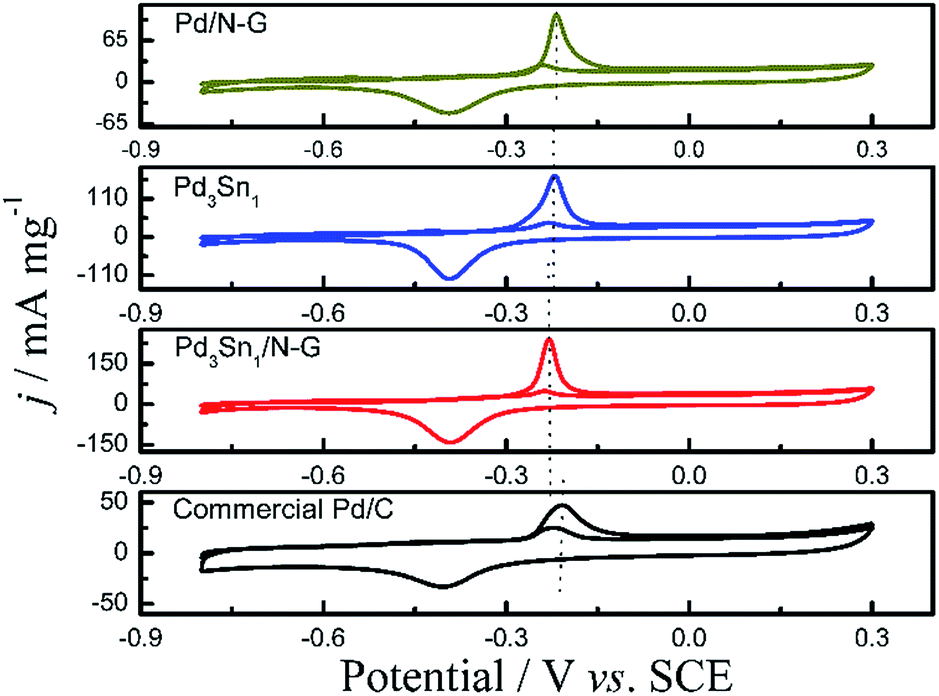
Fig. 5 shows cyclic voltammograms (CVs) of the nitrogen-doped graphene (N-G) supporting Pd and PdSn catalysts, Pd3Sn1 catalyst without nitrogen-doped graphene and commercial Pd/C in an alkaline medium of 1.0 M KOH solution from −0.8 V to 0.3 V at a scan rate of 50 mV s−1. For all these as-prepared catalysts, a typical redox peak at approximately −0.4 V was observed in the cathodic scan which was associated with palladium oxide. In the anodic scan shown in Fig. 5A, all the PdSn/N-G catalysts exhibit positive shifting of peak potentials compared to Pd/N-G, while all PdSn–NRGO catalysts also show more positive shifting of the cathodic peak potentials in the cathodic scans. The distinct anodic (oxidation) and cathodic (reduction) behaviors on PdSn/N-G and Pd/N-G catalysts indicate that the electronic structure on the surface of Pd–Sn has changed after modified with Sn. The electrochemical active surface area (ECSA) of the catalysts was determined from the coulombic charge for the reduction of palladium oxide, according to eqn (1).37
| |
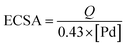 | (1) |
[Pd] in mg represents the palladium loading in the electrode;
Q in mC represents reduction charge of the PdO that was measured by integration of the area under the CV reduction peak; the constant 0.43 (mC cm
−2) was assumed for the reduction of the PdO monolayer. In
Fig. 5, the Pd
3Sn
1/N-G catalyst presents the highest reduction peak current density. As shown in
Table 2, the Pd
3Sn
1/N-G catalyst provided the largest ECSA (562.02 cm
2 mg
−1), higher than other catalysts. The higher ECSA value of PdSn/N-G was most likely owing to the presence of more active sites, which significantly enhanced its electrocatalytic activity for alcohol oxidation.
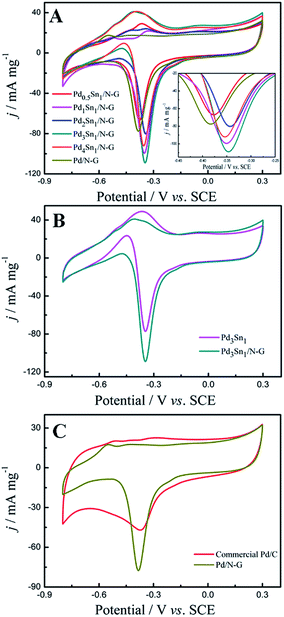 |
| | Fig. 5 Cyclic voltammograms of Pd0.5Sn1/N-G, Pd1Sn1/N-G, Pd2Sn1/N-G, Pd3Sn1/N-G, Pd4Sn1/N-G and Pd/N-G (A); Pd3Sn1 and Pd3Sn1/N-G (B); commercial Pd/C and Pd/N-G (C) in 1.0 M KOH solution at a scan rate of 50 mV s−1. | |
Table 2 Catalytic performance of various Pd–Sn catalysts for ethanol oxidation in alkaline solution
| Samples |
ECSA (cm2 mg−1) |
If (mA mg−1) |
| Pd0.5Sn1/N-G |
237.24 |
2015.00 |
| Pd1Sn1/N-G |
387.62 |
2193.75 |
| Pd2Sn1/N-G |
336.24 |
2173.33 |
| Pd3Sn1/N-G |
562.02 |
2983.33 |
| Pd4Sn1/N-G |
407.95 |
2422.92 |
| Pd/N-G |
329.46 |
2047.92 |
| Pd3Sn1 |
457.36 |
2479.17 |
| Commercial Pd/C |
109.01 |
643.00 |
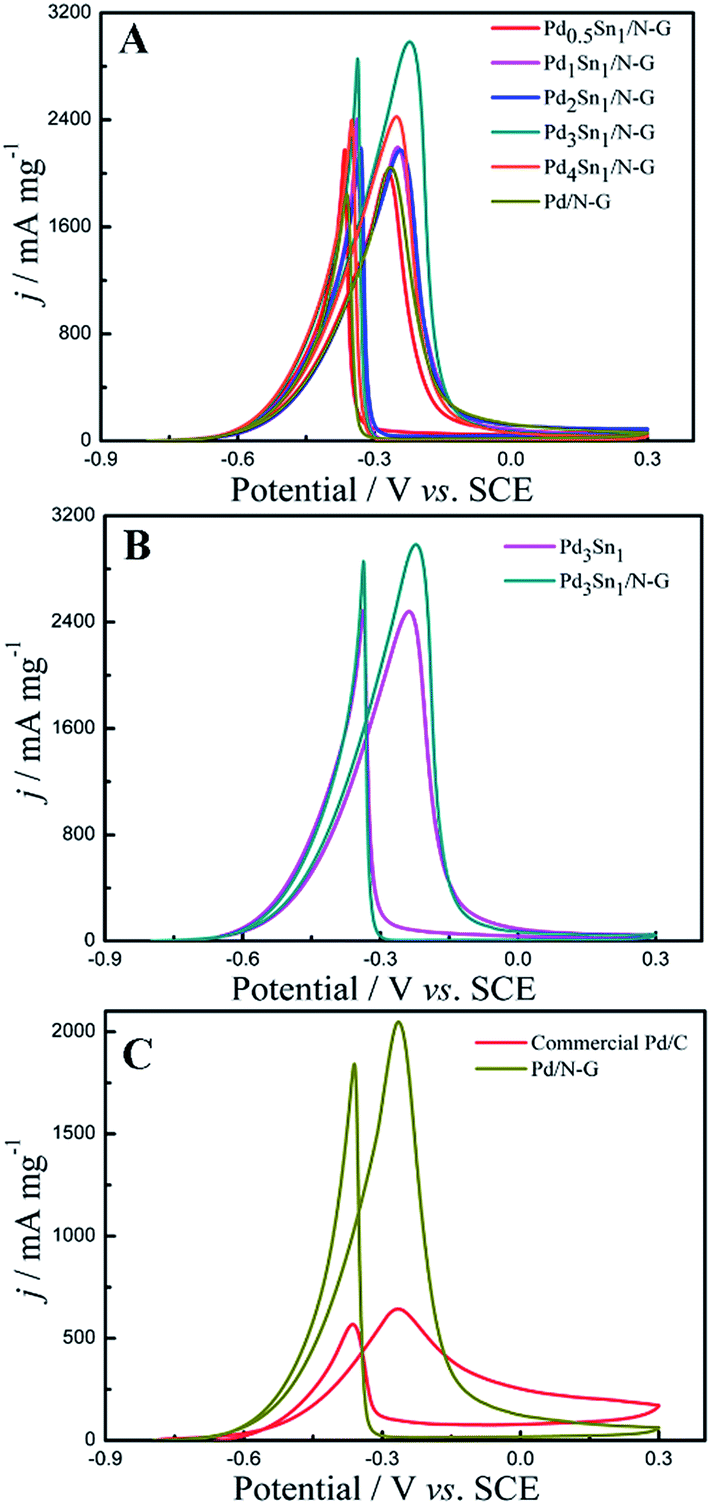
The electrochemical catalytic activities of the as-prepared catalysts were measured by cyclic voltammetry (CV) in an alkaline solution containing 1.0 M C2H5OH and 1.0 M KOH, and the CV results are recorded in Fig. 6. As seen in Fig. 6, two oxidation peaks are presented in the CV grams during the scan: the peak in the forward sweep at about −0.25 V corresponds to the direct oxidation of freshly chemisorbed species coming from ethanol adsorption, and the other peak in the backward sweep at about −0.40 V is related to the removal of carbonaceous species not completely oxidized in the forward scan.38,39 The current density is normalized to the amount of loaded Pd. It can be observed that the forward peak current densities were 2015 mA mg−1, 2193.75 mA mg−1, 2173.33 mA mg−1, 2983.33 mA mg−1, 2422.92 mA mg−1, 2047.92 mA mg−1, 2479.17 mA mg−1, 643 mA mg−1 for Pd0.5Sn1/N-G, Pd1Sn1/N-G, Pd2Sn1/N-G, Pd3Sn1/N-G, Pd4Sn1/N-G, Pd/N-G, Pd3Sn1 and commercial Pd/C, respectively. The ethanol oxidation activity of the PdSn/N-G catalyst was compared to others in the literatures. For example, Zhu et al.40 prepared high activity of carbon nanotubes supported PdSn catalysts for ethanol electrooxidation. The PdSn/CNTs catalyst can reach the activity of 791.97 mA mg−1. Modibedi et al.8 reported that, carbon supported Pd–Sn nanocatalysts exhibit high activity with about 1831.67 mA mg−1. PdSn bimetallic catalysts supported on sulfonated MWCNTs were fabricated by Ramulifho et al.41 The electrochemical performance data of ethanol oxidation on SF–MWCNT–PdSnmix catalyst is 717.76 mA mg−1. These results revealed that the N-G is a superior material for PdSn bimetallic catalysts. The results indicate that the Pd3Sn1/N-G catalyst shows the superior activity to other catalysts.
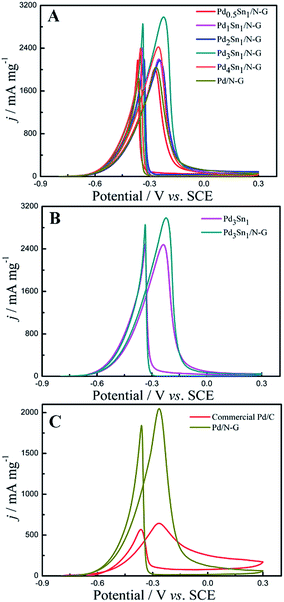 |
| | Fig. 6 Cyclic voltammograms of Pd0.5Sn1/N-G, Pd1Sn1/N-G, Pd2Sn1/N-G, Pd3Sn1/N-G, Pd4Sn1/N-G and Pd/N-G (A); Pd3Sn1 and Pd3Sn1/N-G (B); commercial Pd/C and Pd/N-G (C) in 1.0 M C2H5OH/1.0 M KOH solution. | |
However, it is hard to distinguish the activity promoting is derived from the ECSA or the effect of N-G material. To clarify this point, the ECSA-normalized specific activities of Pd3Sn1 and Pd3Sn1/N-G catalysts are shown in Fig. 7. The ECSA-normalized specific activity of Pd3Sn1 is a little higher than Pd3Sn1/N-G, while the mass-normalized activity of Pd3Sn1 is lower than Pd3Sn1/N-G catalyst in Fig. 6B, indicating that the activity of the as-prepared catalyst is derived from the supporting of N-G.
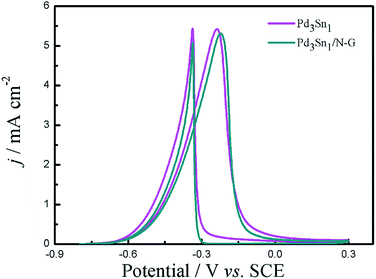 |
| | Fig. 7 Cyclic voltammograms of Pd3Sn1 and Pd3Sn1/N-G in 1.0 M C2H5OH/1.0 M KOH solution at a scan rate of 50 mV s−1. | |
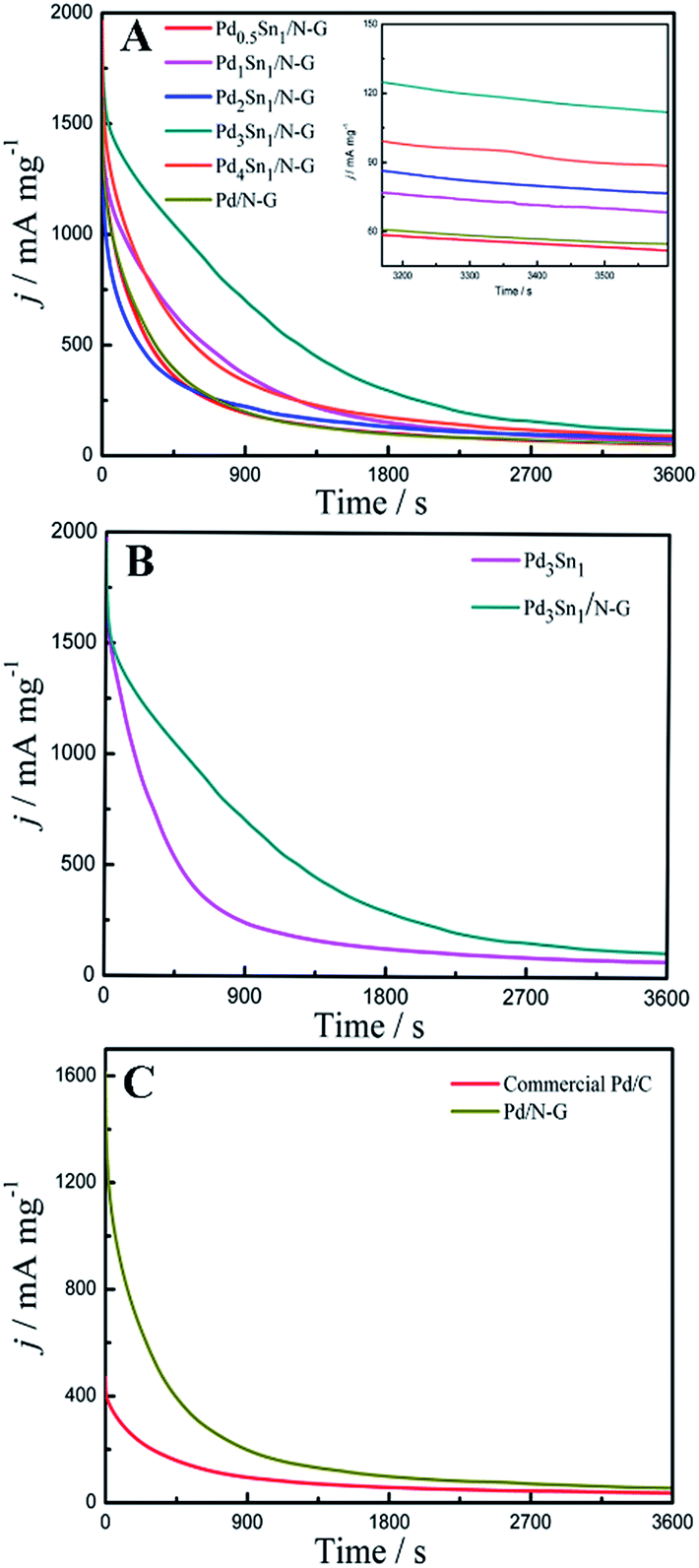
The long-term stability and durability of all these catalysts were further investigated by chronoamperometric measurements in a 1.0 M C2H5OH + 1.0 M KOH solution at −0.3 V (vs. SCE), which was exhibited in Fig. 8. For all the samples, the current density decrease rapidly at the beginning, which is attributed to the poisoning of the catalysts by the intermediate species such as COads formed during the ethanol oxidation reaction.42 After 1800 s, the current density tends to be stable. As shown in Fig. 8A, the residual current density of the Pd3Sn1/N-G catalyst after 3600 s was 115 mA mg−1, which was higher than any other catalysts supported on N-G, indicating that the moderate amount of Sn is helpful to increase the stability of Pd catalysts supported on N-G. When compare Pd3Sn1 and Pd3Sn1/N-G catalysts, Pd/N-G and commercial Pd/C catalysts, higher current densities of Pd3Sn1/N-G and Pd/N-G were also observed in Fig. 8B and C, which mean that N-G is an excellent supporting material for metal catalysts better than carbon. These results suggest that both the addition of Sn and N-G can enhance the stability of Pd catalysts for ethanol oxidation, which probably result from the bimetallic synergy and the strengthened metal support interaction by the incorporation of nitrogen.
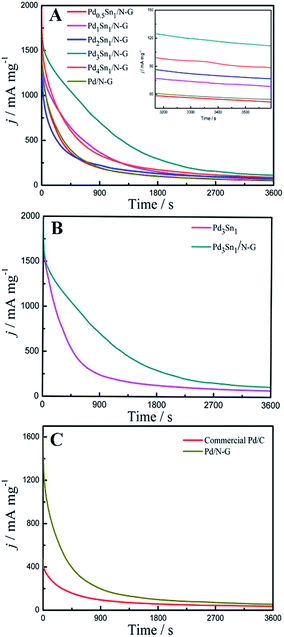 |
| | Fig. 8 Chronoamperograms of Pd0.5Sn1/N-G, Pd1Sn1/N-G, Pd2Sn1/N-G, Pd3Sn1/N-G, Pd4Sn1/N-G and Pd/N-G (A); Pd3Sn1 and Pd3Sn1/N-G (B); commercial Pd/C and Pd/N-G (C) in 1.0 M C2H5OH/1.0 M KOH solution. | |
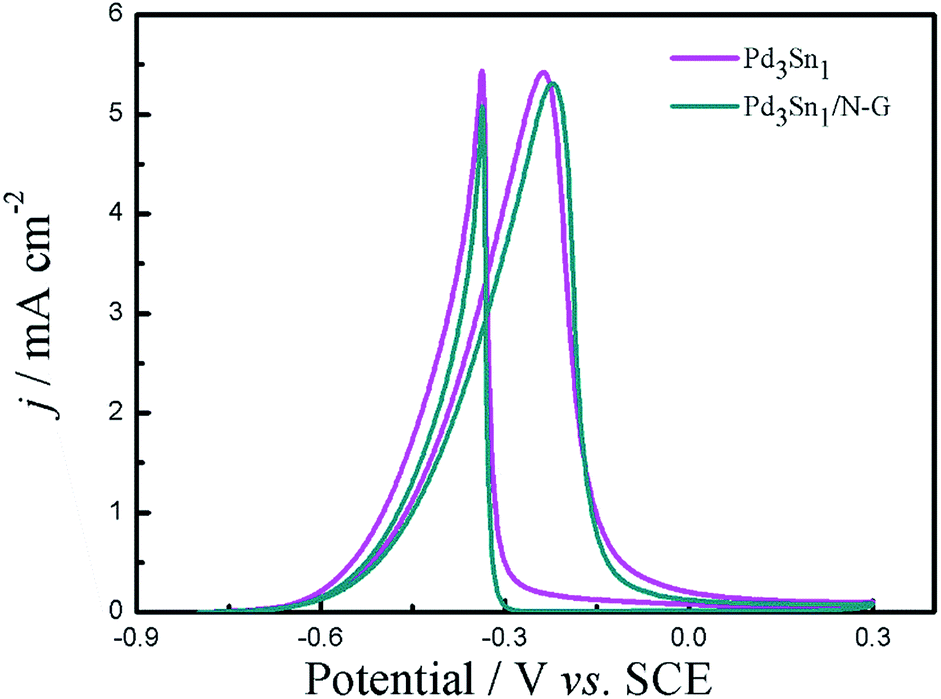
Fig. 9 shows the CVs of CO stripping on the catalysts in 1.0 M KOH solution at a scan rate of 50 mV s−1. For the catalysts Pd/N-G, Pd3Sn1, Pd3Sn1/N-G and commercial Pd/C, an obvious CO oxidation peak was observed in the forward scan, and a peak of Pd reduction was also exhibited in the backward scan.43 The CO oxidation peak in the following scan nearly disappeared indicated the removal of the pre-adsorbed CO on the catalysts.44 The oxidation peak potential of CO is at −0.23 V on the Pd3Sn1/N-G, apparently lower than Pd3Sn1 (−0.22 V), Pd/N-G (−0.21 V) and commercial Pd/C (−0.20 V) catalysts, which means that the CO adsorbed on the Pd3Sn1/N-G catalyst is more easily removed and oxidized at a lower potential. In other words, Pd3Sn1/N-G catalyst possesses higher tolerance to CO poisoning to ethanol oxidation compared with other catalysts, which is in consistent with the previous result (Fig. 9).
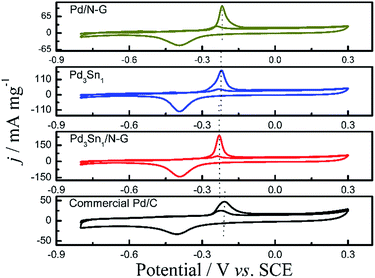 |
| | Fig. 9 CVs of CO stripping on the catalysts in 1.0 M KOH solution, scan rate: 50 mV s−1. | |
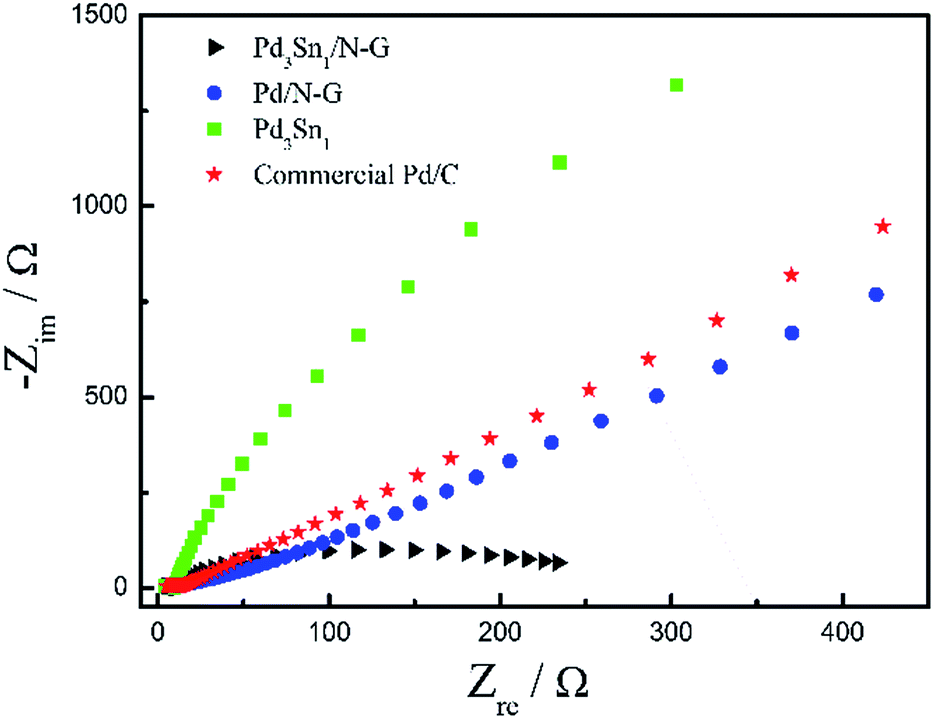
Fig. 10 exhibits the Nyquist plots of electrochemical impedance spectroscopy (EIS) of Pd3Sn1/N-G, Pd/N-G, Pd3Sn1 and commercial Pd/C catalysts in 1.0 M C2H5OH/1.0 M KOH solution at electrode potential −0.25 V, which is an efficient measurement to determine the charge transfer resistance. As known, the diameter of the semicircle is used to measure the resistance of the catalysts, and the smaller diameter of the impedance arc (DIA) means the smaller charge transfer resistance of the catalysts for ethanol electrochemical oxidation.28 The semicircle diameter of Pd3Sn1/N-G was smaller than Pd/N-G, Pd/C and Pd3Sn1, suggesting that Pd3Sn1/N-G catalysts had smaller charge transfer resistance, further demonstrating that N-G can improve the charge transfer kinetics.45 Many researchers have found that nitrogen is an excellent element for the chemical doping of carbon materials because of its comparable atomic size and contains five valence electrons available to form strong valence bonds with carbon atoms. And their studies also show that N-doped graphene has different properties compared with the pristine graphene, such as the spin density, charge distribution of carbon atoms and a better electron transfer efficiency.46,47 Combine the CV results with the EIS results, the electrochemical activity of Pd3Sn1/N-G and Pd3Sn1 catalyst for ethanol oxidation reaction was close, while the charge transfer resistance of Pd3Sn1/N-G is lower than Pd3Sn1 catalyst. The results show that the conductivity of the Pd3Sn1 nanoparticles containing N-G on the surface may be better than Pd3Sn1 catalyst without N-G supporting, indicating that the introduction of nitrogen into graphene can improve the transfer kinetics of metal catalysts.
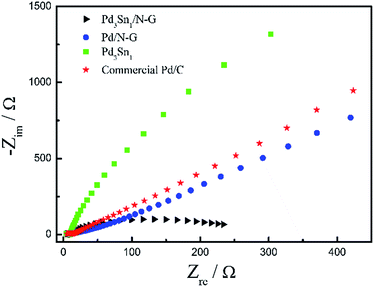 |
| | Fig. 10 Nyquist plots of ethanol electrooxidation on Pd3Sn1/N-G, Pd/N-G, Pd3Sn1 and commercial Pd/C in 1.0 M C2H5OH/1.0 M KOH solution at electrode potential −0.25 V. | |
Conclusions
In this work, the PdSn composite catalysts with different molar ratio were successfully fabricated by a facile chemical reduction method using NaBH4 as a reducing agent and the nitrogen-doped graphene was used as a support material for PdSn catalysts. The as-prepared catalysts displayed network structure and dispersed well with small size. The electrocatalytic performance of all these catalysts were investigated compared with commercial Pd/C. According to the results of CV, CA and EIS, Pd/Sn with the molar ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, supported by nitrogen-doped graphene exhibited higher catalytic activity and stability toward ethanol oxidation than Pd0.5Sn1/N-G, Pd1Sn1/N-G, Pd2Sn1/N-G, Pd4Sn1/N-G, Pd/N-G, Pd3Sn1 and commercial Pd/C, indicating that the moderate amount of Sn and the support of nitrogen-doped graphene can enhance the catalytic properties of Pd catalysts.
:1, supported by nitrogen-doped graphene exhibited higher catalytic activity and stability toward ethanol oxidation than Pd0.5Sn1/N-G, Pd1Sn1/N-G, Pd2Sn1/N-G, Pd4Sn1/N-G, Pd/N-G, Pd3Sn1 and commercial Pd/C, indicating that the moderate amount of Sn and the support of nitrogen-doped graphene can enhance the catalytic properties of Pd catalysts.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51373111), Suzhou Nano-project (ZXG2012022), State and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Notes and references
- E. Antolini, J. Power Sources, 2007, 170, 1–12 CrossRef CAS.
- W. Du, G. Yang, E. Wong, N. A. Deskins, A. I. Frenkel, D. Su and X. Teng, J. Am. Chem. Soc., 2014, 136, 10862–10865 CrossRef CAS PubMed.
- Q. Zhang, J. Zheng, T. Xu, A. Wang, J. Wei, J. Chen and J. Feng, Electrochim. Acta, 2014, 132, 551–560 CrossRef CAS.
- H. Mao, L. Wang, P. Zhu, Q. Xu and Q. Li, Int. J. Hydrogen Energy, 2014, 39, 17583–17588 CrossRef CAS.
- H. Wang, Z. Liu, Y. Ma, K. Julian, S. Ji, V. Linkov and R. Wang, Phys. Chem. Chem. Phys., 2013, 15, 13999–14005 RSC.
- W. Z. Hung, W. H. Chung, D. S. Tsai, D. P. Wilkinson and Y. S. Huang, Electrochim. Acta, 2010, 55, 2116–2122 CrossRef CAS.
- L. X. Ding, A. L. Wang, Y. N. Ou, Q. Li, R. Guo, W. X. Zhao, Y. X. Tong and G. R. Li, Sci. Rep., 2013, 3, 1181 Search PubMed.
- R. M. Modibedi, T. Masombuka and M. K. Mathe, Int. J. Hydrogen Energy, 2011, 36, 4664–4672 CrossRef CAS.
- D. Tu, B. Wu, B. Wang, C. Deng and Y. Gao, Appl. Catal., B, 2011, 103, 163–168 CrossRef.
- D. He, Y. Jiang, H. Lv, M. Pan and S. Mu, Appl. Catal., B, 2013, 132–133, 379–388 CrossRef CAS.
- D. He, K. Cheng, H. Li, T. Peng, F. Xu, S. Mu and M. Pan, Langmuir, 2012, 28, 3979–3986 CrossRef CAS PubMed.
- E. Antolini, Appl. Catal., B, 2009, 88, 1–24 CrossRef CAS.
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, J. Power Sources, 2011, 196, 4873–4885 CrossRef CAS.
- Z. Wu, W. Ren, L. Gao, J. Zhao, Z. Chen, B. Liu, D. Tang, B. Yu, C. Jiang and H. Cheng, ACS Nano, 2009, 3, 411–417 CrossRef CAS PubMed.
- Y. Shao, M. F. El-Kady, L. J. Wang, Q. Zhang, Y. Li, H. Wang, M. F. Mousavi and R. B. Kaner, Chem. Soc. Rev., 2015, 44, 3639–3665 RSC.
- L. Gao, W. Yue, S. Tao and L. Fan, Langmuir, 2013, 29, 957–964 CrossRef CAS PubMed.
- S. Chen, J. Duan, J. Ran, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2013, 6, 3693 CAS.
- J. Mei and L. Zhang, Electrochim. Acta, 2015, 173, 338–344 CrossRef CAS.
- A. K. Singh, S. Jang, J. Y. Kim, S. Sharma, K. C. Basavaraju, M. G. Kim, K. R. Kim, J. S. Lee, H. H. Lee and D. P. Kim, ACS Catal., 2015, 5, 6964–6972 CrossRef CAS.
- M. Prabu, P. Ramakrishnan and S. Shanmugam, Electrochem. Commun., 2014, 41, 59–63 CrossRef CAS.
- J. Bai, Q. Zhu, Z. Lv, H. Dong, J. Yu and L. Dong, Int. J. Hydrogen Energy, 2013, 38, 1413–1418 CrossRef CAS.
- H. An, H. Cui, D. Zhou, D. Tao, B. Li, J. Zhai and Q. Li, Electrochim. Acta, 2013, 92, 176–182 CrossRef CAS.
- D. Bin, B. Yang, F. Ren, K. Zhang, P. Yang and Y. Du, J. Mater. Chem. A, 2015, 3, 14001–14006 CAS.
- D. Bin, F. Ren, H. Wang, K. Zhang, B. Yang, C. Zhai, M. Zhu, P. Yang and Y. Du, RSC Adv., 2014, 4, 39612 RSC.
- N. Cheng, H. Lv, W. Wang, S. Mu, M. Pan and F. Marken, J. Power Sources, 2010, 195, 7246–7249 CrossRef CAS.
- F. Colmati, E. Antolini and E. R. Gonzalez, Appl. Catal., B, 2007, 73, 106–115 CrossRef CAS.
- T. I. T. Okpalugo, P. Papakonstantinou, H. Murphy, J. McLaughlin and N. M. D. Brown, Carbon, 2005, 43, 153–161 CrossRef CAS.
- X. Xu, Y. Zhou, J. Lu, X. Tian, H. Zhu and J. Liu, Electrochim. Acta, 2014, 120, 439–451 CrossRef CAS.
- B. Choi, H. Yoon, I. S. Park, J. Jang and Y. E. Sung, Carbon, 2007, 45, 2496–2501 CrossRef CAS.
- B. Xiong, Y. Zhou, Y. Zhao, J. Wang, X. Chen, R. O'Hayre and Z. Shao, Carbon, 2013, 52, 181–192 CrossRef CAS.
- K. Zhang, D. Bin, B. Yang, C. Wang, F. Ren and Y. Du, Nanoscale, 2015, 7, 12445–12451 RSC.
- T. S. Almeida, A. R. Van Wassen, R. B. VanDover, A. R. de Andrade and H. D. Abruña, J. Power Sources, 2015, 284, 623–630 CrossRef CAS.
- G. Li and P. G. Pickup, J. Power Sources, 2007, 173, 121–129 CrossRef CAS.
- D. H. Lim, D. H. Choi, W. D. Lee and H. I. Lee, Appl. Catal., B, 2009, 89, 484–493 CrossRef CAS.
- H. Wang, X. Qiao, J. Chen, X. Wang and S. Ding, Mater. Chem. Phys., 2005, 94, 449–453 CrossRef CAS.
- F. Lan, D. Wang, S. Lu, J. Zhang, D. Liang, S. Peng, Y. Liu and Y. Xiang, J. Mater. Chem. A, 2013, 1, 1548–1552 CAS.
- J. Yang, Y. Xie, R. Wang, B. Jiang, C. Tian, G. Mu, J. Yin, B. Wang and H. Fu, ACS Appl. Mater. Interfaces, 2013, 5, 6571–6579 CAS.
- T. Maiyalagan and K. Scott, J. Power Sources, 2010, 195, 5246–5251 CrossRef CAS.
- R. N. Singh and A. Singh, Carbon, 2009, 47, 271–278 CrossRef CAS.
- F. Zhu, G. Ma, Z. Bai, R. Hang, B. Tang, Z. Zhang and X. Wang, J. Power Sources, 2013, 242, 610–620 CrossRef CAS.
- T. Ramulifho, K. I. Ozoemena, R. M. Modibedi, C. J. Jafta and M. K. Mathe, Electrochim. Acta, 2012, 59, 310–320 CrossRef CAS.
- C. W. Kuo, L. M. Huang, T.-C. Wen and A. Gopalan, Power Sources, 2006, 160, 65–72 CrossRef CAS.
- R. Yue, H. Wang, D. Bin, J. Xu, Y. Du, W. Lu and J. Guo, J. Mater. Chem. A, 2015, 3, 1077–1088 CAS.
- Y. Zhao, L. Zhan, J. Tian, S. Nie and Z. Ning, Electrochim. Acta, 2011, 56, 1967–1972 CrossRef CAS.
- H. Wang, C. Zhang, Z. Liu, L. Wang, P. Han, H. Xu, K. Zhang, S. Dong, J. Yao and G. Cui, J. Mater. Chem., 2011, 21, 5430 RSC.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- D. W. Wang, I. R. Gentle and G. Q. Lu, Electrochem. Commun., 2010, 12, 1423–1427 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.