
Ni nanoparticles highly dispersed on ZrO2 and modified with La2O3 for CO methanation
Abstract
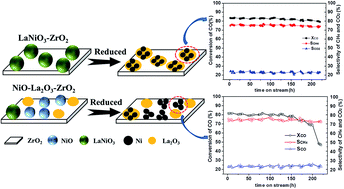
To improve the anti-sintering and anti-carbon deposition ability of the supported metallic nano catalysts, a new scheme for designing and preparing catalysts for CO methanation is presented in this work. In the scheme, a series of xLaNiO3/ZrO2 (x = 15%, 20%, 25%) catalysts were prepared according to the citrate complexing method. The catalysts were characterized by using BET, XRD, H2-chemisorption, H2-TPR, and TEM technologies. After reduction, the LaNiO3/ZrO2 catalyst precursor preferred to form Ni nanoparticles highly dispersed on ZrO2 and modified with La2O3. Compared with the catalyst prepared by the traditional impregnation method, the Ni/La2O3–ZrO2 derived from LaNiO3/ZrO2 showed a higher dispersion of Ni on ZrO2 and exhibited higher CO conversion and CH4 selectivity. Meanwhile, the LaNiO3/ZrO2 catalyst showed excellent stability owing to the significant improvement in both anti-carbon deposition and anti-sintering. The catalyst prepared according to this scheme intensified the interaction between Ni and La2O3, thus favoring the synergistic effect of La2O3 and Ni, which led to the high dispersion of Ni nanoparticles as well as the very good anti-sintering and anti-carbon deposition ability.
 Please wait while we load your content...
Please wait while we load your content...

