
A facile, one-pot procedure for the conversion of aromatic aldehydes to esters, as well as thioesters and amides, via acyl hydrazide intermediates†
Abstract
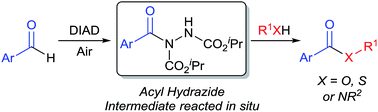
Herein we present an efficient method for the synthesis of esters from aromatic aldehydes via readily accessible acyl hydrazides. The developed reaction protocol is shown to be tolerant of a range of aromatic aldehydes, bearing various functionalities, as well as being amenable to the synthesis of thioesters and amides.
 Please wait while we load your content...
Please wait while we load your content...

