
Electronic and optical properties of surface hydrogenated armchair graphene nanoribbons: a theoretical study†
Abstract
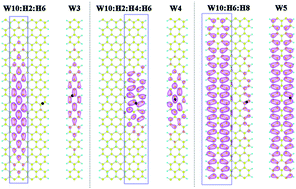
The electronic and optical properties of surface hydrogenated armchair graphene nanoribbons (H-AGNRs) are investigated by first-principle ab initio calculations with quasi-particle corrections. The variation in band gaps is scrutinized in terms of bonding characteristics and the localization of wavefunctions. Optical absorption spectra, exciton binding energies and exciton wavefunctions are investigated with the consideration of different hydrogen adsorption row positions and coverages. Instead of the traditional family effect in pristine AGNRs, we introduce an effective width model to provide a more general understanding for H-AGNRs. The calculations show that the effective width segment in H-AGNRs plays an important role in the band gaps and excitons. Moreover, the spatial distributions of the electronic and exciton wavefunctions are confined by hydrogen atoms, revealing a self-confinement pseudo quantum well.
 Please wait while we load your content...
Please wait while we load your content...

