
Predicting the reducing power of organic super electron donors†
Abstract
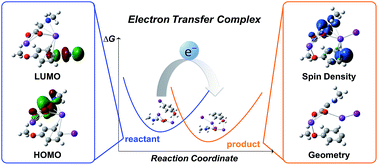
The utilization of computational methods to predict reactivity is an increasingly useful tool for chemists to save time and materials by screening compounds for desirable reactivity prior to testing in the laboratory. In the field of electron transfer reactions, screening can be performed through the application of Marcus Hush theory to calculate the activation free energy of any potential reaction. This work describes the most accurate and efficient approach for modelling the electron transfer process. In particular, the importance of using an electron transfer complex to model these reactions rather than considering donor and acceptor molecules as separate entities is highlighted. The use of the complex model is found to produce more accurate calculation of the electron transfer energy when the donor and acceptor spin densities are adequately localised.
 Please wait while we load your content...
Please wait while we load your content...

