
Solid-solution alloying of immiscible metals at the nanoscale: Ir and Au†
Abstract
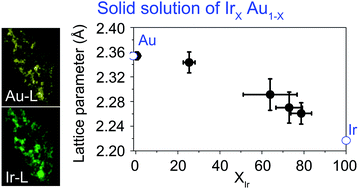
A solid solution alloy of Ir–Au has been theoretically predicted to be an excellent catalyst for dissociating hydrogen, equivalent to Pt. However, Ir and Au atoms are known to be thermodynamically immiscible. In this work, we show that the thermodynamics of the Ir and Au binary system can be changed by nanoscale effects and that electrochemically synthesized Ir–Au alloys have a solid-solution structure with a wide range of compositions. This work is expected to open up the potential synthesis of new catalytic materials.
 Please wait while we load your content...
Please wait while we load your content...

