
Progress in the production of biomass-to-liquid biofuels to decarbonize the transport sector – prospects and challenges
Abdul Waheed
Bhutto
ab,
Khadija
Qureshi
a,
Rashid
Abro
c,
Khanji
Harijan
d,
Zheng
Zhao
c,
Aqeel Ahmed
Bazmi
e,
Tauqeer
Abbas
e and
Guangren
Yu
*c
aDepartment of Chemical Engineering, Mehran University of Engineering and Technology, Jamshoro 76062, Pakistan
bDepartment of Chemical Engineering, Dawood University of Engineering & Technology, Karachi, Pakistan
cBeijing Key Laboratory of Membrane Science and Technology & College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China. E-mail: gryu@mail.buct.edu.cn; Fax: +86-10-6443-3570; Tel: +86-10-6443-3570
dDepartment of Mechanical Engineering, Mehran University of Engineering and Technology, Jamshoro 76062, Pakistan
eProcess and Energy Systems Engineering Center-PRESTIGE, Department of Chemical Engineering, COMSATS Institute of Information Technology, Lahore, Pakistan
First published on 17th March 2016
Abstract
Annually the transport sector consumes a quarter of global primary energy and is responsible for related greenhouse gas emissions. Presently, petroleum derived liquid fuels are the overwhelming source of energy for the transport sector. Liquid biofuels are a viable substitution for petroleum-derived fuels in the transport sector and an important option to mitigate greenhouse gas emissions, especially CO2 emissions. Substituting petroleum-derived fuels with liquid biofuel is also anticipated to reduce the dependency of the transport sector on fossil fuels. Different options are available for the production liquid biofuels. However, the production of liquid biofuels from lignocellulosic biomass has certain advantages. These advantages include the high abundance, availability, low procurement cost and current under-utilization of lignocellulosic biomass. However, the potential for successful deployment of technologies to produce liquid biofuel from lignocellulosic biomass and their cost reductions are surrounded by large uncertainties. High cost of production of liquid fuels from lignocellulosic biomass and their commercial immaturity are major obstacles for the widespread application of liquid biofuels in transportation. Other obstacles include the lack of infrastructure and lack of political as well as public support. This article reviews the obstacles behind the limited production of biomass to liquid (BTL) fuels and their diffusion in the transport sector. The potential approaches to make the production of lignocellulosic-based liquid biofuels economically attractive are also discussed. An approach that focuses on integrating individual operations and processes and adequately modelling these processes evaluated on the bases of the entire pathway can help in realizing the large scale commercial production of liquid biofuels through cleaner production.
 Abdul Waheed Bhutto | Abdul Waheed Bhutto is an Assistant Professor at Dawood University of Engineering & Technology, Karachi. He is currently a PhD candidate at Mehran University Jamshoro. He is concentrating his efforts on improving the efficiency of processes for biofuel production from lignocellulosic material. He received his Master's Degree from NED University Karachi in 2001. He earned his Bachelor's degree in chemical engineering from Dawood Engineering College Karachi in 1998. His research work has focused on alternate energy sources for sustainable development. He is an author of more than 30 research papers. |
 Khadija Qureshi | Dr Khadija Qureshi is a Professor at Mehran University-Jamshoro. She has an extensive research and teaching background in chemical and environmental engineering. For 15 years she has supervised research projects on the reuse of waste materials and agricultural waste, the conversion of agricultural waste into useful chemicals and liquid fuels. She was also a post doctorate fellow at the University of Arizona working on the US Science and Technology Program. She is also a key trainer in gender and mainstreaming women in leadership and management. |
 Rashid Abro | Rashid Abro is a Chemical Engineer at the Pakistan Council of Scientific and Industrial Research (PCSIR) in Pakistan. He is a currently PhD scholar (funded by Higher Education Commission – HEC, Pakistan) at Beijing university of Chemical Technology under the supervision of Dr Xiaochun Chen. His PhD research focuses on the applications of ionic liquids for production of clean fuels. He received his Bachelors and Master's Degrees in Chemical Engineering in 2007 and 2011, respectively, from Mehran University of Engineering and Technology, Jamshoro, Pakistan. He also worked as a lecturer in the Department of Chemical Engineering, Dawood University of Engineering and Technology, Karachi for three years. |
 Khanji Harijan | Dr Khanji Harijan is a Professor at Mehran University-Jamshoro. He received his PhD from Mehran University-Jamshoro in 2008 where he worked on the modeling and analysis of the potential demand for renewable sources of energy in Pakistan. He has an extensive research and teaching background in renewable energy. He is the author of over 100 articles and book chapters. His current research focuses on identifying and addressing barriers to renewable energy development. |
 Aqeel Ahmed Bazmi | Dr Aqeel Ahmed Bazmi is a professional chemical engineer and holds PhD (Chemical Engineering) degree from UTM, Malaysia. He has been served as Lecturer in Chemical Engineering at Dawood College of Engineering & Technology (DCET), Karachi for seven years and joined the Department of Chemical Engineering, COMSATS Institute of Information Technology (CIIT), Lahore in 2006 as Assistant Professor. He is an author of several high impact factor articles in reputed ISI-indexed international journals. His research areas are sustainable development; environmental modeling; bioprocess engineering; optimization modeling; process system engineering. |
 Guangren Yu | Dr Guangren Yu is a professor in the College of Chemical Engineering, Beijing University of Chemical Engineering (BUCT). He received his BS, MS and PhD degrees in chemical engineering from Shandong University of Industrial Technology in 2000, BUCT in 2003 and Institute of Process Engineering, Chinese Academy of Sciences in 2007, respectively. In 2012, he did one-year visiting research at the University of California at Berkeley, USA under the supervision of Professor John M. Prausnitz. He is an author of more than 50 research papers and patents. He is researching on the theory and application of ionic liquids in clean fuel production, biofuels, acid gas capture separation, computer simulation and QSPR. |
1. Introduction
Almost 95 per cent of the world's transportation energy comes from petroleum-based fuels, largely gasoline and diesel. The transport sector is also responsible for 14 per cent of global greenhouse gas (GHG) emissions and around 25 per cent of energy-related CO2 emissions.1–3 Energy use in transport sector and related carbon emissions are projected to be about 80 per cent higher than current levels by 2030.4 Due to reliance on fossil fuels and persistent demand, transport sector is quite difficult to decarbonize. According to the projections by Pietzcker and co-workers,5 in first half of the century, transport sector de-carbonization lags 10–30 years behind mitigation efforts in the non-transport sectors in all models when subject to the same monetary incentives to decarbonize. Only Brazil has achieved success in replacing petroleum fuels in the transport sector with liquid biofuel.6 Decarbonizing the transport sector is thus a fundamental challenge that needs to be tackled to limit climate change.Conventional transportation fuels are composed of petroleum based liquid hydrocarbons predominantly gasoline and diesel. The gasoline contains carbon numbers between C5–C12 mostly branched structures while the carbon numbers of diesel is between C10–C20 mostly linear structures.7 The entire transportation infrastructure has been developed to base upon the properties of petroleum-based liquid fuels.7 These fuels possess high energy-density, stability and superior combustion characteristics. All these properties are highly desired for transportation fuels.7
Shifting dependence from petroleum to renewable biomass resources is important development towards the sustainable development and effective management of GHG. Various technologies are under development for converting biomass to chemicals and transportation fuels. Next generation biofuels are those that rely on lignocellulosic material more generally known as biomass. Each year, more than 40 × 109 kg of inedible plant material is produced and most of which is thrown away.8 Turning these discarded woody bits of plants into ‘next-generation biofuels’ has also huge appeal of not taking food away from a hungry planet.7,8 Domestic production of liquid biofuel and its utilization as alternative transportation fuel can also help to reduce dependency on petroleum oil, reduce air pollution and reduce CO2 emissions.9 Promotion of domestic production of liquid biofuels from indigenous resources provide the opportunity for non-oil-producing countries to lesser their dependence on oil import.10 According to International Energy Agency (IEA) projection, biofuels could provide 27% of total transport fuel by 2050. The projected use of biofuels could avoid around 2.1 × 1012 kg CO2 emissions per year when produced sustainably.11Fig. 1 shows the major benefits of biofuels.12
 | ||
| Fig. 1 The major benefits of biofuels. | ||
There are number of major conversion pathways for the production of liquid fuels from biomass.13 An overview of the pathway for production of biofuels from biomass is summarized in Fig. 2. The selection of biomass feedstock depend on cropping pattern, crops yields, area under consideration, local conditions, food coproduction, economics, and the life cycle energy efficiency.
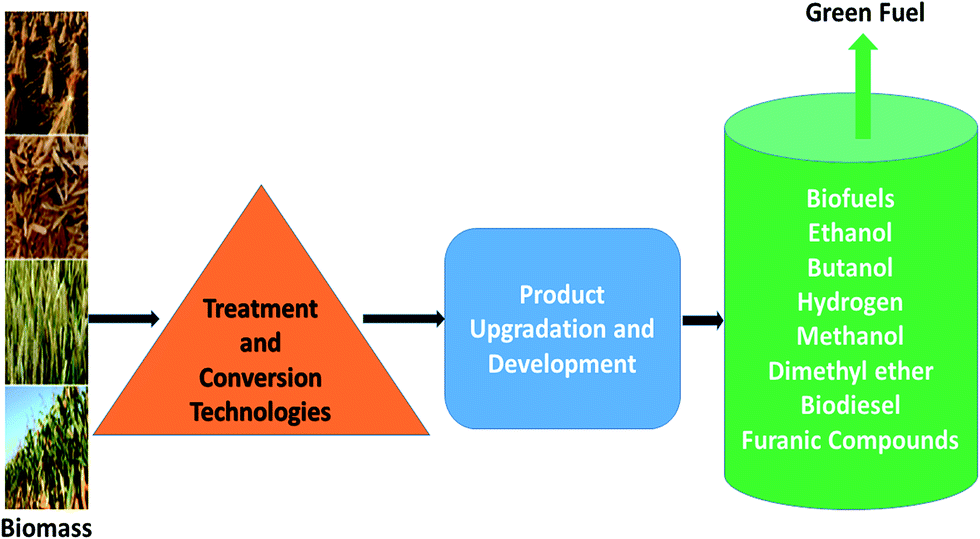 | ||
| Fig. 2 The complete pathway for production of biofuels from biomass. | ||
2. Biomass resources and basic conversion technologies
2.1 Biomass resources
Biomass stores sunlight in the form of chemical energy through photosynthesis. When biomass is processed to extract energy stored in it, carbon is oxidized to produce CO2 and water. The process is cyclical, as the CO2 produced during oxidation can convert back to fresh biomass through reduction reactions. Table 114 gives a detailed category of various kinds of biomass resources and examples for every type.| Categories | Representative materials | ||
|---|---|---|---|
| Productive biomass | Terrestrial | Carbohydrate | Sugar cane, corn, sweet sorghum |
| Starch | Maize, cassava, sweet potato | ||
| Cellulose | Tropical grasses, poplar, sycamore | ||
| Hydrocarbon | Eucalyptus, green coral | ||
| Grease | Oil palm, rapeseed, sunflower | ||
| Aquatic | Freshwater | Water hyacinth | |
| Ocean | Large kelp | ||
| Microorganism | Green algae, photosynthetic bacteria | ||
| Unused biomass | Residues from agriculture forestry fisheries | Agriculture | Wheat bran, straw, vegetable residues, processing residues |
| Animal husbandry | Animal manure | ||
| Farm residues | |||
| Forestry | Secondary forest, woodland remnants, damaged plant material | ||
| Waste | Fisheries | Jettisoned and dead fish | |
| Municipal waste | Municipal and pulp sludge | ||
| Garbage | Family garbage, feces | ||
2.2 Strategies for production of fuels from biomass
Biomass can be converted into next generation liquid biofuels by three primary routes. The first route is the gasification of biomass to produce synthetic gas, which is subsequently converted into liquid fuels. Second route is the pyrolysis or liquefaction of biomass to produce liquid bio-oil. Third route is the hydrolysis of biomass to produce sugar monomer units. Liquid bio-oils produced through above three routes must be upgraded if they are to be used as transportation fuels. The overall strategy in the production of liquid hydrocarbon fuels from biomass is (i) to break existing C bonds with different compounds (ii) to create C–C bonds (iii) to increase the molecular weight (iv) to remove oxygen from the product and (iv) to improve energy density.152.3 Basic conversion technologies
The production of first generation biofuels is based upon well-established technologies. However with the development of these biofuels a competition was developed between the biofuel and the food industries over feedstock availability. This competition shift the focus from producing first generation biofuels towards successful production of commercially viable next-generation biofuels to take advantage availability of large volume and variety of feedstock. In general, lignocellulosic feedstocks are converted into next-generation liquid biofuels through following distinct conversion routes also illustrated in Fig. 3. | ||
| Fig. 3 Simplified schematic illustration of the two main biofuel production pathways. | ||
(i) Thermochemical conversion of biomass to syngas followed by catalytic conversion into hydrocarbons directly associated with liquid fuel mixtures.
(ii) Thermo-chemical conversion of biomass into complex liquid product followed by upgrading inferior liquid bio-oil to fuel quality liquid fuel.
(iii) Thermochemical conversion of biomass to syngas followed by syngas fermentation to produce liquid biofuels.
(iv) Enzyme hydrolysis of biomass into sugars (e.g. glucose and xylose) followed by liquid phase fermentation.
2.4 Thermochemical conversion
Thermochemical methods of conversion utilizes the entire feedstock and eliminate the energy-intensive pretreatment steps.16 Pre-treatment step prior to gasification consists only of screening, size reduction, wet storage, drying and dry storage.17 These methods are feedstock-flexible and products are produced in a relatively shorter span of time.18 Thermochemical conversion and subsequent product upgradation are key technologies for production of next generation liquid biofuels and a pathway for sustainable development. However, the multiple interactions processing steps greatly increase the difficulty in the accurate design of such processes.19 Hence this promising route for the production next generation liquid biofuels is still not fully adopted on large scale.| Operating procedure | Characteristics |
|---|---|
| (a) Updraft fixed bed | |
| Operation | Characteristics |
| • Can handle larger particle sizes (20–100 mm) and a wider range of moisture contents (5–55%) | • High concentration of tar compounds in the raw syngas |
| • The biomass is fed in at the top of the gasifier while the oxidizing gas enters the reactor at bottom, hence the biomass and gases move in countercurrent directions | • Temperature of syngas at reactor exit is about 250 °C which is exploited to dry biomass off and the system sensitivity to biomass moisture content is less |
| • Char burning provide the heat to reactor | • It can handle biomass with moisture content up to 50% |
| • The raw syngas leaves through top of the gasifier while unburned char and ash falls from the grate for collection at the bottom of the gasifier | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| (b) Downdraft fixed bed | |
| Operation | Characteristics |
| • Can handle larger particle sizes (20–100 mm) and a wider range of moisture contents (5–55%) | • Low concentration of tar compounds in the raw syngas |
| • The biomass is fed in at the top of the gasifier. Oxidizing gas or steam intake is also at the top or from the sides | • Syngas temperature at its exit from the reactor is about 800 °C |
| • Biomass and oxidizing gas move partly in the concurrent direction in bottom section | • Sensitive to the quality of biomass feedstock. Moisture content in range between 10 and 25% is required |
| • Part of biomass burns and falling through the gasifier throat to form a bed of hot charcoal though which the gases have to pass through (a reaction zone). This ensures a fairly high quality syngas, which leaves at the base of the gasifier while ash is collected under the grate | |
|
|
| (c) Entrained flow | |
| Operation | Characteristics |
| • Feedstock particle size (0.1–1 mm), moisture content (<15 wt%) and a constant composition | • Preparing fine size feedstock require pre-treatment (fine grinding, drying) which consequent high cost |
| • Powdered biomass is fed into a gasifier with pressurized oxygen and/or steam from the top of reactor | • Very high operating temperatures (>1200 °C) and pressures (up to 40 and 50 bars) |
| • A turbulent flame at the top of the gasifier burns some of the biomass to provide large amounts of heat at high temperature around 1200–1500 °C | • At high temperature ash melts, cools down, and eventually accumulates as slag. Hence reactor is not suitable for high ash content feedstocks |
| • High temperature at top section ensure fast conversion of biomass into very high quality syngas | • Very high oxygen demand |
| • The ash melts onto the gasifier walls, and is discharged as molten slag | • Short residence time |
| • Requires sophisticated reactor design and construction materials | |
|
|
| (d) Bubbling fluidized bed (BFB) | |
| Operation | Characteristics |
| • A bed of fine inert material sits at the gasifier bottom, while oxidizing gas blown upwards through the bed at very high velocity (1–3 m s−1) to agitate the material | • Uniform temperature distribution |
| • Biomass is fed in from the side which mixes and combusts or forms syngas which leaves upwards | • Intensive particle circulation due to bubble movement promotes good gas and solid mixing |
| • Reactor operates at temperatures below 900 °C to avoid ash melting and sticking. Reactor can be pressurized | • Low reaction temperature ensure low emissions of nitrogen oxides |
| • No slagging | |
|
|
| (e) Circulating fluidized bed (CFB) | |
| Operation | Characteristics |
| • A bed of fine inert material has oxidizing gas blown upwards through the bed fast enough (5–10 m s−1) to suspend material throughout the gasifier | • A clean process with the ability to achieve lower emissions |
| • Biomass is fed in from the side. Part of it combusts to provide heat while remaining biomass reacts to form syngas | • Medium operating temperature (850 °C) |
| • The mixture of syngas and particles are separated using a cyclone, with solid material returned into the base of the gasifier | • Feed circulation rate is control by the gas velocity in the bed which determines the flow regime and density of bed |
| • Perfect gas–solid mixing | |
|
|
| (f) Dual fluidized bed (dual FB) | |
| Operation | Characteristics |
| • This system has two chambers – a gasifier and a combustor | • Operates at temperatures below 900 °C to avoid ash melting and sticking |
| • Biomass is fed into the CFB/BFB gasification chamber, and converted to nitrogen-free syngas and char using steam | • Could be pressurized |
| • The char is burnt in air in the CFB/BFB combustion chamber, heating the accompanying bed particles | |
| • This hot bed material is then fed back into the gasification chamber, providing the indirect reaction heat | |
| • Cyclones remove any CFB chamber syngas or flue gas | |
|
|
| (g) Plasma | |
| Operation | Characteristics |
| • Biomass feedstock with light or without pre-treatment can be supplied to a plasma gasifier | • Usually reactor operates at atmospheric pressure and very high temperatures of 1500–5000 °C |
| • Untreated biomass is dropped into the gasifier where it comes into contact with electrically generated plasma | • Achieve zero-waste |
| • Biomass is converted into very high quality syngas, and inorganic matter is vitrified into inert slag | |
| • Plasma gasification uses plasma torches | |
| • It is also possible to use plasma arcs in a subsequent process step for syngas clean-up | |
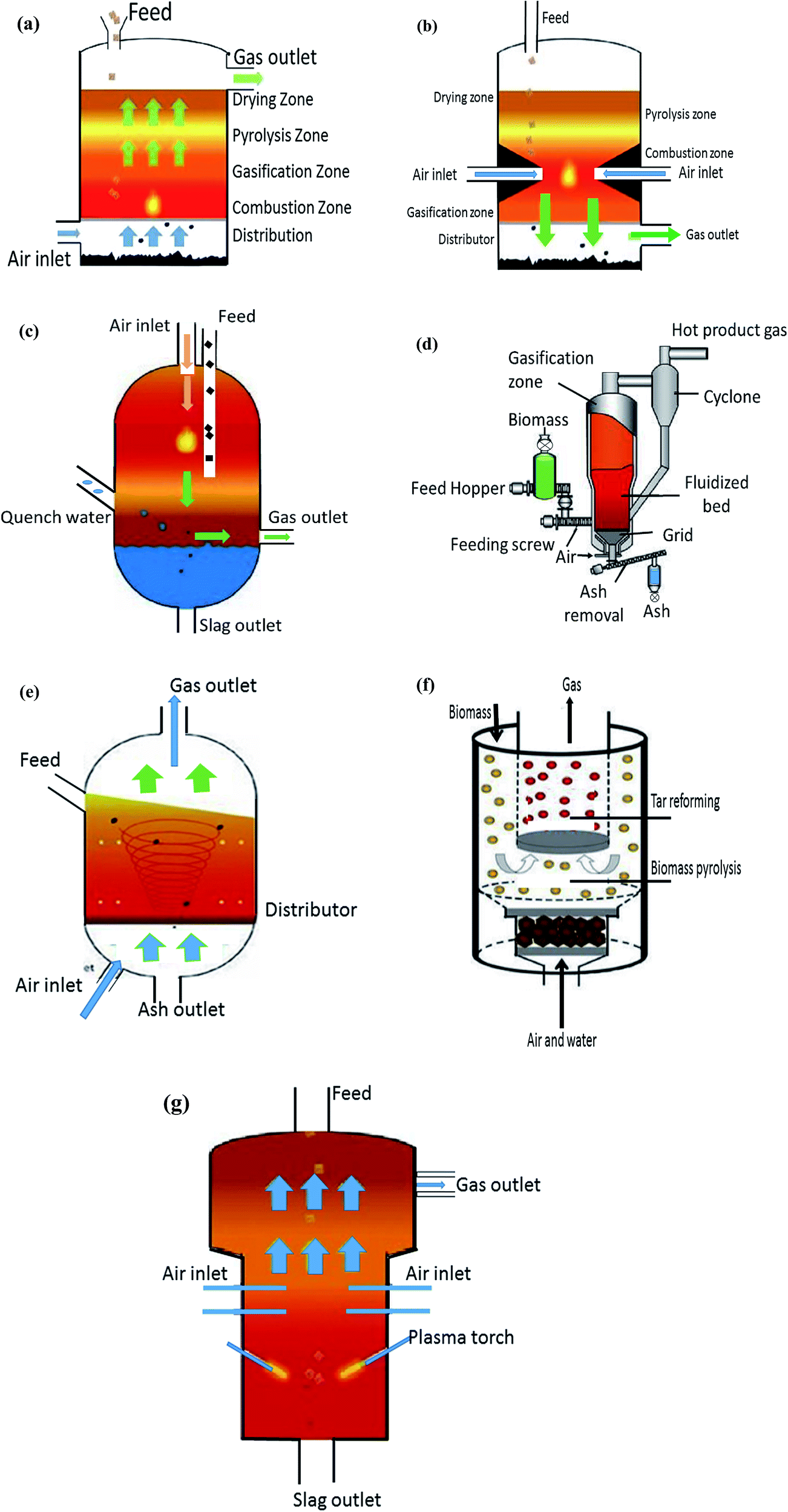 | ||
| Fig. 4 Operating diagrams of gasifiers (a) updraft fixed bed (b) downdraft fixed bed (c) entrained flow (d) bubbling fluidized bed (BFB) (e) circulating fluidised bed (CFB) (f) dual fluidized bed (dual FB) and (g) plasma. | ||
2.4.1.1 Feedstock materials and gasification reactions. Gasification offers a high flexibility in using different kind of feedstock materials as well as in the generation of different products through upgrading the syngas.23Table 324 shows fundamental reactions and enthalpy of selected cellulose gasification reactions.
| Classification | Stoichiometry | Enthalpy (kJ g−1 mol−1) (ref temp 300 K) |
|---|---|---|
| Pyrolysis (thermal decomposition) in absence of oxygen or steam | C6H10O5 → 5CO + 5H2 + C | 180 |
| C6H10O5 → 5CO + CH4 + 3H2 | 300 | |
| C6H10O5 → 3CO + CO2 + 2CH4 + H2 | −142 | |
| Partial oxidation | C6H10O5 + ½O2 → 6CO + 5H2 | 71 |
| C6H10O5 + O2 → 5CO + CO2 + 5H2 | −213 | |
| C6H10O5 + 2O2 → 3CO + 3CO2 + 5H2 | −778 | |
| Steam reforming | C6H10O5 + H2O → 6CO + 6H2 | 310 |
| C6H10O5 + 3H2O → 4CO + 2CO2 + 8H2 | 230 | |
| C6H10O5 + 4H2O → 3CO + 3CO2 + 9H2 | 64 | |
| Water–gas shift reaction | CO + H2O → CO2 + H2 | −41 |
| Methanation | CO + 3H2 → CH4 + H2O | −206 |
2.4.1.2 Biomass gasification products. Raw “producer gas” or “syngas” from biomass gasification is composed of mainly CO, H2, CO2, CH4, N2, water vapor, tars, particulate matter and other trace gases.25 The composition and properties of the syngas varies with type of biomass feedstock, particle size, the gasifier type and the operation conditions of the gasifier. The operating parameters such as type of oxidizing agent, the temperature and the residence time in the gasifier defines the operating conditions. Typical composition of syngas from biomass gasification are given Table 4.
| Constituents | % by volume (dry and nitrogen free) |
|---|---|
| Carbon monoxide (CO) | 28–36 |
| Hydrogen (H2) | 22–32 |
| Carbon dioxide (CO2) | 21–30 |
| Methane (CH4) | 8–11 |
| Ethene (C2H4) | 2–4 |
| Benzene–toluene–xylene (BTX) | 0.84–0.96 |
| Ethane (C2H5) | 0.16–0.22 |
| Tar | 0.15–0.24 |
| Others (NH3, H2S, HCl, dust, ash etc.) | <0.021 |
Main impurities in the syngas are fly ash particles and tar.23 Measures to overcome tar related problems can be classified into primary and secondary. Primary measures focus on the development of better gasifier designs to reduce the production of tars within the gasifier. These include the appropriate selection of operating parameters, the proper design of the gasifier and the use of suitable bed additives or catalysts during gasification.27 As a primary measure De Filippis et al.28 has proposed gasifier reactor followed by a secondary fixed bed reactor filled with aluminum oxide spheres having high porosity dedicated to the tar conversion reactions. The second reactor has demonstrated a high efficiency in tar removal, with a decrease of more than 50%.28 Secondary measures focus on the destruction or filtration of tars downstream of the gasifier.25 About 40–99%, tar can be reduced by the different secondary measures such as water scrubber, venturi scrubber, cyclone, ESP and rotational particle separator. These methods are discussed in detail by Han and Kim.29 Other impurities in the syngas are typically H2S, cabonyl sulphide (COS), HCl, alkenes, and ammonia.30 Biomass also contains potassium, sodium, and other alkali that can cause slagging and fouling problems in conventional gasification equipment.
The production of syngas from biomass allows the production of different liquid fuel. Pathways for different thermochemical liquid fuel production from syngas are shown in Fig. 5.22
 | ||
| Fig. 5 Pathways for fuel production from syngas.22 | ||
The fuels produced from syngas includes H2 by the water–gas shift reaction, methanol by methanol synthesis, alkanes by FTS, isobutane by isosynthesis, ethanol by fermentation, or with homogeneous catalysts and aldehydes or alcohols by oxosynthesis.31 Synthesis of Fischer–Tropsch (FT) diesel, dimethyl ether (DME), methanol and methane are established processes.23
Pyrolysis is generally categorized as slow, moderate and fast. Slow pyrolysis mainly produce charcoal. Fast and flash pyrolysis at high temperatures with very short residence times convert biomass to a maximum quantity of bio-oil.26,35,36 Fast pyrolysis proceeds by rapid heating of biomass to moderate temperature of 500 °C in the absence of oxygen and immediate quenching of the emerging pyrolysis vapours. Once biomass reaches 300 °C, it thermally depolymerizes to form small oxygenates which are vapours in the reactor but liquid mixture at room temperature. Fast pyrolysis is the most feasible way to convert biomass into liquid fuels and give highest yield to liquid fuel products and retains most of the energy from feedstock.37,38 Catalytic fast pyrolysis combines the fast pyrolysis of biomass with the catalytic transformation of the primary pyrolysis vapors to more desirable and less oxygenated liquid fuels. These liquid fuels can readily be upgraded to transportable liquid while simultaneously increasing energy density.39,40,41 Operating parameters like temperature, heating rate, residence time, and particle size all affect bio-oil yield as well as its quantity.39
2.4.2.1 Pyrolysis products and impurities. Depending on the type of feedstock, pyrolysis produces 60–75% liquid bio-oil, 15–25% solid char and 10–20% non-condensable gases.32,42 The chemical composition of the bio-oil vary sharply according to the type of biomass. Bio-oil produced during the pyrolysis of biomass is dark colored, viscous and unstable liquid. It contains the complex mixture of more than 300 oxygenated compounds mainly includes acids, alcohols, aldehydes, esters, ketones, phenols, and lignin-derived oligomers. It also contains considerable water content (15–30 wt%).43 The low heating value and flame temperature, greater ignition delay, and lower combustion rate of bio-oil are largely due to the high water content. The physical properties of bio-oil such as density, acidity, viscosity and chemical compositions change during storage and transportation, which is one of the most challenging problems in using bio-oil for any applications including as transportation fuels.44 The adverse characteristics of bio-oil particularly the instability of bio-oil is associated with the high oxygen content in the bio-oil. These oxygenates also causes many other negative properties, such as low heating value, high corrosiveness and high viscosity. Oxygen content also greatly limit the application of bio-oil as transportation fuel.45 Upon storage bio-oil undergo series of chemical reactions triggered by organic acids and intermediates in addition to the re-polymerization of reactive olefinic compounds. The chemical composition of bio-oil classified by functional groups is shown in Fig. 6.31 Bio-oil produced through pyrolysis contains (i) small carbonyl compounds such as acetic acid, acetaldehyde, acetone, hydroxyaldehydes, hydroxyketones, and carboxylic acids; (ii) sugar-derived compounds such as furfural, levoglucosan, anhydrosugars, furan/pyran ring-containing compounds; and (iii) lignin-derived compounds, which are mainly phenols and guaiacols.39 Bio-oil also contains oligomers of a molecular weight ranging from 900 to 2500 in significant quantity.39 Because of complex compositions bio-oils show a wide range of boiling temperatures. Fig. 716 shows that bio-oil composition in terms of carbon-to-oxygen ratio and yield vary significantly depending on feedstock type, inorganic content and reaction conditions.16
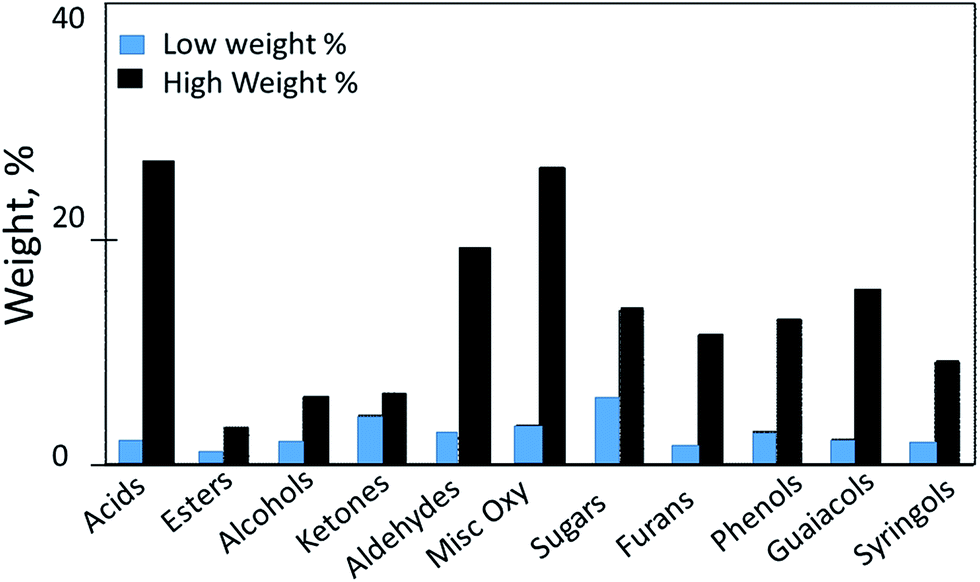 | ||
| Fig. 6 Chemical composition of bio-oil from biomass and the most abundant molecules of each of the components.31 | ||
 | ||
| Fig. 7 Bio-oil composition (carbon-to-oxygen ratio) vs. yield for various feedstocks based on a weight average of all volatile products (do not include char or permanent gases (CO and CO2)). The reaction temperature is 500 °C unless noted otherwise16 [reprinted from ref. 16 with permission of Royal Society of Chemistry]. | ||
Bio-oil is distinctly dissimilar to petroleum-derived oils. Summaries of the characteristics of pyrolysis oil and fuel oil are shown in Table 5.
| Property | Pyrolysis bio-oil | Liquefaction bio-oil | Heavy fuel oil |
|---|---|---|---|
| a at 50 °C, b at 61 °C | |||
| Carbon [wt%] | 54–58 | 73 | 85 |
| Hydrogen [wt%] | 5.5–7.0 | 8 | 11 |
| Oxygen [wt%] | 35–40 | 16 | 1.0 |
| Nitrogen [wt%] | 0–0.2 | — | 0.3 |
| Ash [wt%] | 0–0.2 | — | 0.1 |
| Moisture content [wt%] | 15–30 | 5.1 | 0.1 |
| pH | 2.5 | — | — |
| Specific gravity | 1.2 | 1.1 | 0.94 |
| Higher heating value [MJ kg−1] | 16–19 | 34 | 40 |
| Viscosity [cP] | 40–100a | 15000b |
180a |
| Solids [wt%] | 0.2–1 | — | 1 |
| Distillation residue [wt%] | Up to 50 | — | |
2.4.2.2 Pyrolysis catalyst. Solid acids such as zeolites, silica–alumina, silicalite, FCC catalysts, alumina, molecular sieves, as well as metal oxides such as zinc oxide, zirconia, ceria, and copper chromite have been studied as catalysts in catalytic cracking of pyrolysis vapors.39 Catalyst lower the reaction temperature and improve the selectivity to the desired products.47 Proper selection of catalyst significantly alter the bio-oil composition by promoting deoxygenation, cracking, and reforming reactions. The ideal catalyst should be cheap, strong, stable, resistant to coke formation, regenerable, and effective in terms of its activity and selectivity.47
HTL process has advantageous that no drying of biomass is required compared to pyrolysis and gasification processes which usually require energy intensive drying process prior to pyrolysis treatment.50Table 6 compares technique feasibility of bio-oil production by the flash pyrolysis and HTL and the product characteristic. During HTL, lower process temperature and presence of solvent dilutes the concentration of products and prevents formation of tar compounds.51,52 Employing different solvents could change the distribution and concentration of compounds produced.51
| Methods | Technique feasibility | Challenges | |
|---|---|---|---|
| Pros. | Cons. | ||
| Flash pyrolysis | (i) High oil yield up to 80% on dry feed basis | (i) Poor fuel quality | (i) Improvement of the reliability of pyrolysis reactors and processes36 |
| (ii) The demonstration of the oil's utilization in boilers, engines and turbines36 | |||
| (ii) Lower capital cost | |||
| (iii) The development of technologies for the production of chemicals and biofuels from pyrolysis oils36 | |||
| (iii) Already commercialized | |||
| HTL | (i) Produces improved quality bio-oil with high heating value and low moisture content | (i) Relatively low oil yield (20–60%) | (i) Better understanding of the reaction mechanisms and kinetics |
| (ii) Require high pressure equipment's which increases capital cost | (ii) Improvement of oil production rate and energy efficiency of the process | ||
| (iii) Development of pilot-scale plant and subsequent commercialization of HTL | |||
Review by Huang and Yuan53 offer an important reference for the study of biomass liquefaction with reference to (i) effect of solvent type on the liquefaction behaviours of biomass; (ii) the effect of biomass type on the liquefaction behaviours of biomass; (iii) the liquefaction of biomass in sub-/super-critical ethanol; the liquefaction of biomass in organic solvent–water mixed solvents and (v) the liquefaction of sewage sludge.
2.5 Chemical/biochemical route
Biochemical conversion of biomass into energy fuel is one among the few routes which provide environment sustainable and economically viable solution.| (C6H10O5)n + nH2O → nC6H12O6 |
2.5.1.1 Enzymatic hydrolysis. Variety of microorganisms including bacteria and fungi contain enzymes with the ability to hydrolyze cellulosic biomass to glucose monomers. These enzymes are naturally occurring proteins.54 During enzymatic hydrolysis of lignocellulosic biomass, utilization of both the cellulose and the hemicellulose part is desirable which requires well-designed cocktail of enzymes. The organisms with broad substrate ranges and cellulolytic and/or hemicellulolytic abilities generally suffer from poor growth characteristics or poor product-producing characteristics. The chemical and physical recalcitrance of lignocellulosic biomass also necessitate high enzyme loading to obtain reasonable hydrolysis rates.55 The complete lignocellulose-to-ethanol process using enzymes consists of pretreatment to improve access, enzymatic hydrolysis of cellulose and hemicellulose to sugar monomers, fermentation of sugars to ethanol and ethanol upgradation. The hydrolysis and fermentation is carried out by different enzymes. No organism can directly hydrolyze biomass and ferment the liberated sugar to ethanol at rate and titer required for economic viable operation.56,57 The factors like type of biomass, cellulase activity of enzyme and operating conditions including temperature and pH influences both rate as well as yield during enzymatic hydrolysis.58 Enzymes are intrinsically expensive and thermodynamically unstable. Selection of proper combination of pretreatment and enzymes for a given biomass improves both process efficiency and economic and reduce time requirement.59
Literature estimates for the cost of enzymes varies in the range between $0.10 per gal,60 $0.30 per gal,61 $0.32 per gal,62 $0.35 per gal,63 and $0.40 per gal.64 Reducing the cost of cellulase enzyme production is a key issue in the enzymatic hydrolysis of lignocellulosic materials.54 Since enzymes production is expensive, optimization of enzymatic dosage and recycling of enzymes is necessary for economically feasible ethanol production from biomass.65,66 Significant improvement in the commercially available cocktails of enzymes has been achieved which need further performance evaluation on commercial scale.67 Different commercial organizations including Novozymes and Genencor are working to reduce the production cost of commercial enzymes mixtures and improves the effectiveness of enzymes for biofuel production.55 US government is sponsoring and subsidizing such research. Their activity focuses to (i) reduce unit cost of enzyme by process and strain enhancement and (ii) improvement in the cellulase enzyme performance to reduce its consumption during hydrolysis.68
2.5.1.2 Dilute acid hydrolysis (DAH). Acid hydrolysis is generally of two types: dilute acid hydrolysis (DAH) and concentrated acid hydrolysis (CAH). In DAH the acid strength is generally ≤10% (w/w), whereas in the CAH, the acid concentration is ≥10% (w/w).69 DAH is carried out at 100–240 °C to hydrolyze both cellulose and hemicellulose present in biomass. Since hemicellulose hydrolyzes at 110–140 °C while hydrolysis of cellulose takes place at 240 °C, carrying out the DAH in two stages provide opportunity to take advantage of the differences in hydrolysis temperature of hemicellulose and cellulose.70 Summary of the challenges, advantages of DAH is given in Table 7. Study by Lenihan71 established the optimum conditions for acid hydrolysis of hemicellulosic biomass. The study by Kupiainen72 focused on glucose production from cellulose by dilute acid hydrolysis. The study also provides new knowledge of cellulose hydrolysis and glucose decomposition in formic acid. Pre-treatment of biomass prior to hydrolysis can provide significant cost saving due to the lower temperature requirement and less acid consumption.59
| Property | Description |
|---|---|
| A salt | Cation and/or anion quite large |
| Melting point | Preferably ≤100 °C |
| Liquidus range | Usually >200 °C |
| Thermal stability | Usually high |
| Chemical stability | Usually high |
| Viscosity | High, ≤100 cP workable in liquid–liquid extraction |
| Dielectric constant | Implied ≤30 |
| Polarity | High to moderate |
| Specific conductivity | Usually <10 mS cm−1 |
| Molar conductivity | <10 S cm2 mol−1 |
| Electrochemical window | >2 V |
| Solvent and/or catalyst | Excellent for many organic reactions |
| Vapor pressure | Often negligible |
2.5.1.3 Concentrated acid hydrolysis (CAH). CAH uses concentrated H2SO4 followed by a dilution with water to dissolve and hydrolyze or convert the substrate into sugar.73,74 CAH is carried out at low temperatures and atmospheric pressure which gives high sugar recovery efficiency and minimum sugar degradation. Moderate operating condition also allow use of relatively low cost materials. The energy requirements is also very low. During CAH about 90% of both hemicellulose and cellulose sugar polymers are hydrolyzed into their monomeric fractions. Shahbazi and Zhang has provided the details of both DAH and CAH the apparatus in used and unit operations pertinent to the ethanol industry.75 The acid hydrolysis of hemicelluloses is accomplished easier and faster, compared to cellulose, because of amorphous and heterogeneous structure of hemicelluloses and lower degree of polymerization.76 Common acids for the hydrolysis are mineral acids e.g. sulfuric acid (H2SO4), phosphoric acid (H3PO4), nitric acid (HNO3) and hydrochloric acid (HCl) as well as organic acids such as: trifluoroacetic acid (CF3COOH), oxalic acid (HOOC_COOH) and acetic acid (CH3COOH).
The properties of the modern ionic liquids are summarized in Table 7. Practically, number of salts with low melting points is not limited, and a study has determined this number to be of the order of 1 billion.80
ILs are considered as environmental friendly substitute to classical organic solvents and have widespread application in organic synthesis, electrochemistry, catalysis, etc.81 Being broadly applicative in separation technology these have attracted considerable attention. Fig. 8 contains the common cations families of ILs. While common anions include Cl/[AlCl3], N(CN2), Br, Cl, I, NO3, SO4, CF3COO, CF3SO2, BF4, PF6,HSO4, H2PO4, and CF3(SO2)2N.
 | ||
| Fig. 8 Common cations of ILs (a) imidazolium (b) pyridinium (c) quaternary ammonium (d) phosphate (e) triazolium (f) pyrrolidinium (g) piperidinium (h) guanidinium (i) morpholinium (j) azole alkane (k) amino acid (l) sulfonium. | ||
A range of ILs are used as effective cellulose solvents.82 Native cellulose is not sometimes easily soluble in traditional molecular solvents. Cellulosic material are usually dissolved in derivatizing and non-derivatizing solvents with both aqueous and no aqueous media.83–85 Dissolution of cellulose was first studied by Swatloski et al. (2002) in the IL [Bmim]Cl.86 Soon afterwards, a new type of allyl-functionalized imidazolium-based IL [Amim]Cl, was introduced as an influential cellulose solvent.87,88 The patent of Graenacher in 1934 has been reflected to be the first interpretation of dissolving cellulose in an IL type of solution89 but the used solvent, water-free [BzPy]Cl, contained 1–2% dry pyridine and was as such not a pure salt, neither does the pure [BzPy]Cl fit the generally accepted IL definition of having a melting point below 100 °C. During the last decade, cellulose and wood dissolution in ILs and the underlying mechanisms have been extensively studied and reviewed.90–93 Homogeneous functionalization and derivatization of cellulose in ILs solution has also been studied.84,85
A range of ILs are used as effective cellulose solvents.82 These ILs are [Emim]Ac is the mostly studied IL for treating biomass. It is effective for all type of biomass at moderate reaction conditions with high operating equipment compatibility.94 The anionic part of ILs plays a significant role in fixing an IL's knack to dissolve cellulose. ILs containing acetate, chloride and other moderately basic anions disrupt the hydrogen bond network of cellulose and enable its dissolution.95 Homogeneous catalysis, utilizing low concentrations (less than 5%) of mineral acids such as HCl and H2SO4 in [Bmim]Cl IL showed to effectively hydrolyse cellulose with yields of up to 80%.96,97 Sulfonate resins have also been used to catalyze the selective depolymerisation of cellulose dissolved in [Bmim]Cl.98
IL processes for biomass hydrolysis have several attractive features of working with inexpensive chemical catalysts to produce high sugar yields in few hours at just 105 °C. Lignocellulose solubilization by IL also allows processing at high concentrations. IL process avoids the use of hazardous chemicals. However there are certain challenges in large scale commercialization of IL hydrolysis. Efficient ILs are very expensive in comparison to other solvents. ILs trends to inactivate cellulose.99 Highly viscous biomass–IL mixtures might require special handling. Separation and recycling of IL could pose additional challenges to commercialization. To replace expensive ILs with those derived from renewable sources, Socha et al.100 synthesized a series of tertiary amine-based ILs from aromatic aldehydes derived from lignin and hemicellulose, the major by-products of lignocellulosic biofuel production.
The techno-economic analysis of ILs Klein-Marcuschamer et al.101 has determined that the order of importance of variables involved in economic operation of IL hydrolysis is ILs price > biomass loading > recycling rate. Brandt et al.82 has provided the list contains aspects of ILs treatment for further studies. Xu et al.102 have developed high gravity biomass processing with a one-pot conversion technology that includes ionic liquid pretreatment, enzymatic saccharification, and yeast fermentation for the production of concentrated fermentable sugars and high-titer cellulosic ethanol.
2.6 Development in biofuel production pathways
Table 8 summarizes the challenges, advantages and recent developments in different biofuel production pathways. Mushrif et al.118 identifies that multi-scale molecular modeling of liquid phase catalytic reaction mechanisms and developing a fundamental understanding of the role of solvents in biomass processing can assist in establishing the foundation of biomass processing.| Biofuel production pathways | Challenges | Advantages and recent advances |
|---|---|---|
| Gasification | (i) Tar production is a major problem119 | (i) Shen (2015)120 has proposed tar removal by combining adsorption with catalytic conversion using char-based adsorbents/catalysts |
| (ii) Bhaduri et al.25 has proposed tar tolerant homogeneous charge compression ignition engine fueled by impure syngas at intake temperatures above the tar dew point to avoid the condensation of tars and its consequent problems | ||
| (iii) Other option includes integrated gasification, gas cleaning and conditioning in one reactor unit23 | ||
| Pyrolysis | (i) The presence of high content of O2 and H2O deteriorate the quality of liquid bio-fuel121 | (i) Catalytic fast pyrolysis at 300–500 °C can produce liquid hydrocarbons with the oxygen removed in the form of H2O, CO, and CO2. However, a decrease in liquid yield was observed123 |
| (ii) Bio-oil is poorly suited for direct blend with conventional crude oils in petroleum refinery122 | (ii) Chen et al.124 proposed the torrefaction of biomass before pyrolysis and products upgradation to reduce the number of functional groups containing oxygen | |
| (iii) The lack of detailed knowledge about chemistry and transport is major challenge16 | (iii) Other pretreatment methods that can be employed prior to pyrolysis includes hot water treatment, sulfuric acid and ammonium phosphate doping125 | |
| Hydrothermal liquefaction (HTL) | (i) Selection of effective solvents with proper catalysts to further narrow the product distribution and to produce desired compounds is still a challenge | (i) 80% energy recovery from biomass to fuel in HTL is excellent in comparison to other biomass conversion technologies127 |
| (ii) The process cost of biomass liquefaction is very high and making this competitive with petroleum-based fuels is another challenge53 | (ii) The bio-oil produced by HTL of algae resembled that of petroleum crude except for its high nitrogen and oxygen content which requires further treatment before blending with petroleum crude128 | |
| (iii) The use of water as the processing medium results in a large water handling requirement. Recycle and reuse of the water is a major challenge in optimizing design of hydrothermal processes126 | (iii) The use of homogenous alkaline solutions as catalysts can control the product distribution to improve bio-oil yield and lower the nitrogen and oxygen contents128–130 | |
| (v) Further investigations on catalytic hydrothermal liquefaction is required to develop non-precious metal, composite and hydrothermally stable catalysts | ||
| Enzymatic hydrolysis | (i) High costs of both pretreatment and enzymes (one-third of the cost of ethanol production from cellulose) and low rates of hydrolysis are potential drawbacks to enzymatic hydrolysis131 | (i) Pretreatment of lignocellulosic materials significantly enhance the hydrolysis of cellulose |
| (ii) Significant efforts are required to lower cost of enzymes | (ii) Enzymatic hydrolysis is usually conducted at mild conditions (pH 4.8 and temperature 45–50 °C) and the utility cost of enzymatic hydrolysis is low with no significant corrosion problem132 | |
| (iii) Using genetically engineered microorganisms that can convert xylose and/or pentose to ethanol and optimization of the cellulase enzymes and the enzyme loading can significantly improve ethanol production efficiency and reduce the cost of the production54,133 | ||
| Dilute acid hydrolysis (DAH) | (i) DAH has low sugar recovery efficiency of around 50% | (i)Feedstock size reduction facilitate the sugar recovery,135 whoever reducing the feedstock size significantly increases the pre-treatment cost135 |
| (ii) Formation of furfural-type components during DAH decreases the sugar yields and inhibiting biochemical conversion of the sugars into ethanol134 | ||
| Concentrated acid hydrolysis (CAH) | (i) Optimizing sugar recovery and recycling of acid is still a challenging task136–138 | (i) The advantages of CAH includes high sugar yields, low levels of fermentation inhibitors, good fermentability and robustness towards changes in raw material quality134 |
| (ii) CAH has corrosion issues | ||
| (iii) A neutralization of pH is necessary before fermentation. Lime is used to neutralize the acid which produces large quantities of calcium sulfate. The disposal of calcium sulfate results in additional expense73 | ||
| Ionic liquids (ILs) | (i) Recovery of ILs is challenging because fermentable sugars and [EMIM]Cl, have similar solubilities in various solvents139 while IL in the sugar mixture interfere with enzymes functioning during subsequent fermentation8 | (i) ILs such as [Emim][Ac] are environmentally benign and compatible with organisms used for downstream conversion94 |
| (ii) The cost of ionic liquids (ILs) is very high which impediment commercialization of IL hydrolysis94 | (ii) Protic ionic liquids containing the hydrogen sulfate [HSO4]− anion can compete with other pretreatment methods in terms of effectiveness and process cost | |
| (iii)The replacement of [EMIM]Cl by other ILs could help to overcome the extraction issues by even using conventional separations (e.g., distillation and extraction) which leads to significant cost saving94 | ||
| Mechanical extraction | (i) Excess heating time and/or temperature result in a cake with lower nutritional value and oil with lower quality | (i) Simple operation with robust equipment |
| (ii) Can be maintained and operated by semi-skilled supervisors | ||
| (iii) Produces protein rich cake free of chemical impurities140 | ||
| Chemical extraction | (i) The process design requires further understanding of mass transfer kinetics106 | (i) Supercritical fluid extraction with CO2 ensures an innocuous separation process both to human health and to the environment110 |
| (ii) Properties of CO2 can be tuned to enhance selectivity and provide extracts with desirable compositions110 | ||
| Transesterification of vegetable oils | (i) The high viscosity and flash point and low volumetric heating values of biodiesel are major challenges | (i) It is biodegradable and non-toxic |
| (ii) High cost of biodiesel is the major obstacle to its commercialization141 |
3. Product up gradation and development
3.1 Product up gradation
Upgrading of bio-oil is carried out to improve its quality by reducing or removing one or more of its undesirable component. The undesired characteristics or properties of bio-oil are explained in Table 9.40 Czernik and Bridgwater has discussed these properties in details.142 Upgrading of bio-oil to a conventional transport fuel requires full deoxygenation and some conventional refining. Bio-oil can be upgraded by thermochemical-catalytic, chemical and biochemical treatment.| Characteristic | Cause(s) and effect(s) |
|---|---|
| Acidity | Presence of organic acids reduces the pH which effect the catalyst activity and causes corrosion problems |
| Aging | Secondary reactions during storage such as condensation deteriorate product quality. Slow increase in viscosity from secondary reactions also causes phase separation |
| Alkali metals | High ash feed and incomplete solids separation results in presence of high alkali metal in product. Presence of these metals causes catalyst poisoning |
| Deposition of solids in combustion, erosion and corrosion, slag formation all these damage to turbines and engines | |
| Char | Incomplete char separation causes the aging of oil, sedimentation, filter blockage, catalyst blockage, engine injector blockage and alkali metal poisoning |
| Chlorine | Biomass feed contain high amount of chlorine which poison catalyst during upgrading |
| Color | Cracking of biopolymers and presence of char deteriorate color quality |
| Contamination of feed | Poor harvesting practice results in presence soil component in bio-oil which act as catalysts and results in undesired side reactions. It also increase particulate carry over |
| Distillability is poor | Because of the presence of reactive component in product bio-oil cannot be distilled (at maximum 50% distillation). Liquid begins to react at below 100 °C and substantially decomposes above 100 °C |
| Low H:C ratio |
Presence of oxygen compounds results in low H:C ratio. Upgrading to hydrocarbons is more difficult |
| Materials incompatibility | Presence of phenolic and aromatics with good solvent properties results in destruction of seals and gaskets |
| Miscibility with hydrocarbons is very low | Highly oxygenated nature of bio-oil results in low miscibility with any hydrocarbons so integration into a refinery is more difficult |
| Nitrogen | Presence of nitrogen in biomass feed results in nitrogen contamination which result in unpleasant smell, catalyst poisoning during upgrading of bio-oil and formation of NOx during combustion |
| Oxygen content is very high | Presence of oxygen content results in poor stability during storage and non-miscibility with hydrocarbons |
| Phase separation | High feed water, high ash in feed and poor char separation results in phase separation, layering, poor mixing, inconsistency in handling, storage, and processing |
| Smell | Presence of aldehydes and other volatile organics results in objectionable smell |
| Structure | Rapid de-polymerization and rapid quenching of vapors and aerosols results in susceptibility to aging such as viscosity increase and phase separation |
| Sulfur | Sulfur contaminants in results in catalyst poisoning during upgrading |
| Temperature sensitivity | Presence of temperature sensitive component results in incomplete reactions |
| Irreversible decomposition of liquid into two phases >100 °C, irreversible viscosity increase above 60 °C and phase separation above 60 °C | |
| Toxicity | Biopolymer degradation products may results in positive human toxicity |
| Water | Presence of water in bio-oil results in complex effect on viscosity and stability. High water content also lowers heating value, density, stability, and raises pH. It also affects catalysis |
3.2 Fischer–Tropsch's synthesis (FTS)
The production of high quality fuels from biomass by FTS comprises of the four basic steps. (1) Biomass pre-treatment, (2) gasification of the biomass feedstock to synthesis gas (syngas, CO + H2) followed by gas cleaning/conditioning; (3) FTS production and (4) upgrading of the Fischer–Tropsch's liquids to high quality fuels (Fig. 9). | ||
| Fig. 9 Flow scheme for conversion of biomass feedstocks to liquid hydrocarbon fuels17 [reprinted from ref. 17 with permission from Elsevier]. | ||
FTS process was developed by Franz Fischer and Hans Tropsch in the 1920s through which syngas is converted into long chain liquid hydrocarbons. The Fischer–Tropsch's chemical transformation process is expressed through the following set of reactions:
| (2n + 1)H2 + nCO → CnH(2n+2) + nH2O |
The products from FTS are a range of mostly straight chain alkanes ranging from C1 to C50 governed by the chain polymerization kinetics model known as Anderson–Schulz–Flory (ASF) polymerization model. The ASF model is represented by the following equation:143
| Wn = n(1 − α)2αn−1 |
| logCn = log(ln2α) + nlogα |
Cnversus n should give a straight line graphically represented in Fig. 10. It clearly displays the predicted distributions for several products and product ranges of particular interest. The optimal H2:CO ratio of 2:1 is achieved through the water–gas shift reaction. The reaction temperature depends upon the type and quantities of product desired. Low temperature (200–240 °C) or high temperature (300–350 °C) synthesis at a pressure of 20–40 bar is carried out using iron (Fe) or cobalt (Co) catalyst.145 The proper selection of the catalyst and reactor type may be significant for overall product yield and economics. Fischer–Tropsch's products are further upgraded through hydro processing. Gasification of the solid feedstock with H2O is an endothermic reaction which requires high temperatures whereas FTS is an exothermic reaction carried out at temperature around 200–250 °C. Low FTS temperature conditions significantly lower gasification which maximize the yield of long-chain hydrocarbons that can be upgraded to synthetic diesel. Chemical energy and carbon recovery in the hydrocarbon product are reported in the range of 30–50 and 25–45%, respectively.146 The addition of H2 minimizes O2 consumption in gasification and improves product selectivity during synthesis. It also helps in conversion of CO and CO2 during synthesis. Increased CO2 conversion can also be achieved if H2O is removed decreasing limitations for CO2 hydrogenation.146
 | ||
| Fig. 10 Weight fraction of hydrocarbon chains of length n as a function of the growth probability factor α (ref. 144) [reprinted from ref. 144 with permission of The Royal Society of Chemistry]. | ||
FTS produces a range of olefins, paraffin, and oxygenated compounds (alcohols, aldehydes, acids, and ketones). As shown in Fig. 10, gasoline or diesel fuel cannot be made selectively using FTS without producing a large amount of undesired byproducts. However, regardless of the product type, FTS are predominantly linear with high olefinicity. Beside temperature and pressure other process variables that influence the product distribution includes feed gas composition, catalyst type, and promoters.144
Fig. 11 shows the effect of zeolite addition on the selectivity to different hydrocarbons. Hybrid catalysts suppress the formation of hydrocarbons boiling above the gasoline range (C13+) which increases the selectivity to gasoline range products (C5–C12) by about 60–140% depending on the zeolite properties. Fig. 11 also depicts that the hybrid catalyst containing the small crystal size zeolite (FeZN50) are most selective towards gasoline.17 Higher partial pressures of H2 and CO lead to higher liquid selectivity SC5 whereas inert in the syngas will lower partial pressures of H2 and CO, thereby reducing SC5+.17Table 10 shows the detail of Co-based catalyst for the commercial production of Fischer–Tropsch's products.
 | ||
| Fig. 11 Hydrocarbon distribution for Fischer−Tropsch synthesis with Fe–Co–K (base). The effect of zeolite addition on the selectivity to different hydrocarbons at comparable conversions147 [reprinted from ref. 147 with permission from Elsevier]. | ||
| Properties | Methane | Methanol | Dimethyl ether | Ethanol | Gasoline | Diesel |
|---|---|---|---|---|---|---|
| Formula | CH4 | CH3OH | CH3OCH3 | CH3CH2OH | C7H16 | C14H30 |
| Molecular weight (g mol−1) | 16.04 | 32.04 | 46.07 | 46.07 | 100.2 | 198.4 |
| Density (g cm−3) | 0.00072 | 0.792 | 0.661 | 0.785 | 0.737 | 0.856 |
| Normal boiling point (°C) | −162 | 64 | −24.9 | 78 | 38–204 | 125–400 |
| LHV (kJ cm−3) | 0.0346 | 15.82 | 18.92 | 21.09 | 32.05 | 35.66 |
| LHV (kJ g−1) | 47.79 | 19.99 | 28.62 | 26.87 | 43.47 | 41.66 |
| Exergy (MJ L−1) | 0.037 | 17.8 | 20.63 | 23.1 | 32.84 | 33.32 |
| Exergy (MJ kg−1) | 51.76 | 22.36 | 30.75 | 29.4 | 47.46 | 46.94 |
| Carbon content (wt%) | 74 | 37.5 | 52.2 | 52.2 | 85.5 | 87 |
| Sulfur content (ppm) | ∼7–25 | 0 | 0 | 0 | ∼200 | ∼250 |
The Fischer–Tropsch's hydrocarbons can be hydrocracked to form mainly diesel of excellent quality. H2 is added to remove double bonds, after which the Fischer–Tropsch's liquids are cracked catalytically with H2. The overall carbon efficiency of the hydrocracking step is close to 100%. Fischer–Tropsch's liquids are totally free of sulfur and contain very few aromatics. Fischer–Tropsch's diesel, with a very high cetane number can also be used as a blend stock to improve the quality of normal diesel. The Fischer–Tropsch's naphtha has a much lower octane number than ‘normal’ naphtha.
In Germany, the commercialization of the Fischer–Tropsch's technology began in 1936 using coal as syngas source using low-temperature Fischer–Tropsch's technology.149 Techno-economic studies of gasification plants producing Fischer–Tropsch's by Wright and Brown145 suggest that costs range from $0.3 to $1.1 per L of Fischer–Tropsch's diesel produced. A yield of 120 L diesel fuel per metric tonne biomass obtained from wood by FTS of biomass-derived syngas is lower than 320 L ethanol per tonne biomass yield of ethanol from wood reported by a National Renewable Energy Laboratory (NREL)'s process via hydrolysis and fermentation.31 Boerringter150 has predicted that future improvements could allow the yield to increase to 210 L diesel fuel per tonne biomass. Fischer–Tropsch's process is already in commercial use at Sasol South Africa for production of syngas from coal and Shell Malaysia for production of natural gas-derived syngas.17 However commercial biomass-to-liquid (BTL) process has not been completely established.
3.3 Catalytic cracking
Catalytic cracking upgrades the bio-oils a reducing their oxygen content. Atmospheric catalytic cracking do not requires H2 which reduces the operating cost.46 However, with biomass as feedstock catalytic cracking gives poor yields of hydrocarbons with high coke formation.151 Catalytic cracking of biomass produces hydrocarbons (aromatic, aliphatic), water-soluble organics, water, oil-soluble organics, gases (CO2, CO, light alkanes), and coke. The experiments on catalytic cracking of bio-oil have also been carried out with catalysts, such as Al-SBA-15, alumina and Cu.1523.4 Hydro treating
Hydrogenation of bio-oil is carried out to improve the product quality without alteration of the boiling range. The process is carried out at mild conditions typically high pressure (up to 200 bars) and moderate temperature (up to 400 °C) and also requires H2 supply. Full hydro treating gives a naphtha-like product that requires orthodox refining which can be carried out in conventional refinery to derive conventional transport fuels. A typical naphtha to biomass project yield equivalent to 25% by weight or 55% in energy terms in feedstock excluding provision of H2.40 The conventional hydro processing catalysts, such as CoMo/Al2O3 and NiMo/Al2O3 are used most extensively.153 Noble metals (e.g., platinum) and metal sulfides (e.g., molybdenum sulfide) catalyze the reactions of compounds in bio-oils to give water and hydrocarbons.43 NiMo catalyst presented a higher hydrogenation activity especially at low temperatures.154 Patel and Kumar155 have provided the details of reaction mechanism in bio-oil upgrading, process parameters, and the limitations of hydro processing technology.3.5 Steam reforming
In steam reforming bio-oil is converted to CO and H2 through react with steam at high temperatures. Steam reforming is normally accompanied by the water gas shift and the methanation reaction, where the extents of these reactions depend on the operating conditions.156 The general form of steam reforming of aqueous fractions of bio-oils is given by following equation.157| CnHmOk + (n + k)H2O → nCO + (n + m/2 + k)H2 |
The most promising catalyst are Ni, Rh, or Ru. Ni is preferred as it is the cheapest.156 The fluidized bed process use nickel-based catalysts under conditions similar to those for reforming natural gas. The mixtures of basic oxides and Al2O3 are the most promising support materials as these show higher activity and lower carbon formation. Potassium can be used as a promoter as it can increase the activity and reduce the coke formation.156
3.6 Suitable catalysts to upgrade bio-oil
Whatever thermo-catalytic upgrading process is used to upgrade bio-oil it is imperative to find suitable catalysts. The bi-functional catalysts combines the hydrogenation function of active metal with hydrolysis and dehydration of the support.158 Base metals of Ni, Mo, and Co, noble metals of Pt, Rh, and Ru facilitate hydrogenation reactions. Catalysts supported on SiO2, TiO2, ZrO2, zeolites and various mixed oxide have been explored in recent years.158 Bio-oils upgraded by zeolitic catalysts promote the high yield of liquid products.15,159 HZSM-5 with stronger acidity (low Si/Al ratio) can effectively facilitate cracking reaction of heavy components of crude bio-oil to produce the organic distillate fraction with least coke formation.151,160 During bio-oil up gradation catalyst deactivation can be attributed to large extent to condensation reactions among the partially upgraded components of bio-oil. Recently Weber et al.161 used stochastic model to capture the rates of oligomerization of bio-oil.Bio-oil contains some macromolecular compounds such as napthalene, guaiacol, and lignin-derived oligomers. During upgrading, these compounds were easy to cover the active sites or block the pore of the catalyst, such as zeolites, and make the catalyst deactivate. A combination of the large pore dimensions of mesoporous materials with the strong acid sites would be highly advantageous catalytic material. Microporous zeolite while provide additional diffusion pathways for larger molecules and eliminate the possibility of secondary reactions, which enhance the coke formation and consequently catalyst deactivation caused by a slow mass transport to and away from the catalytic center.159
3.7 Challenges, advantages and recent development in thermal upgrading of bio-oil
Table 11 summarizes the challenges, advantages and recent developments in different thermo-catalytic bio-oil upgrading of technologies. Careful study of the challenges advocate the need to conduct number of mathematical and CFD model at different scales, i.e., kinetic, particle and reactor to understanding the effect of different operating conditions.| Bio-oil upgrading of technology | Challenges | Advantages and recent developments |
|---|---|---|
| Fischer–Tropsch's synthesis (FTS) | (i) Through removal of tar, H2S, carbonyl sulfide, NH3, hydrogen cyanide, alkali and dust particles to avoid poisoning of FTS catalyst is still a challenge17 | (i)The increasing temperature increases conversion, reduces wax production but increases the methane yield |
| (ii) Pressure increase is favorable to the reaction below 20 bar (ref. 162) | ||
| (iii) Liquid fuels obtained from FTS process have similar combustion properties as the gasoline and diesel obtained from petroleum sources.113 FT-diesel possess very high cetane number17 | ||
| Catalytic cracking | (i) High coking (8–25 wt%) and poor quality of the fuels are major challenges151 | (i) HZSM-5 zeolite catalyst promote the conversion of oxygen element in heavy oil into CO, CO2 and H2O (ref. 152) |
| (ii) zeolite cracking rejects oxygen as CO2, yielding mainly aromatic hydrocarbons as product but causes extensive coke deposition on the catalyst | (ii) The sequential biomass pyrolysis reactor which consisted of a traditional catalytic pyrolysis followed by the subsequent catalytic cracking support the decomposition of gaseous intermediate has superiority of promoting the liquid yield and improving the fuel quality over the separate processes152,163 | |
| (iii) Catalysts deactivates by the after three times regenerations.163 Choosing proper catalysts is still a challenge | ||
| Hydrotreating | (i) The high H2 consumption in the bio-oil hydro-processing is the main challenge | (i) When only organic fraction of bio-oil after phase separation is hydrotreated, the H2 requirement can be fulfilled by H2 produced during steam reforming the aqueous phase |
| (ii) High pressure also leads to high operational cost159 | ||
| (iii) Another challenge of hydro-processing is to hydrogenate the aliphatic compounds whilst avoiding reduction of aromatics which is difficult to achieve at high H2 pressure159 | ||
| (iv) Some of the oxygen compounds in the feed readily polymerize to cause fuel instability | ||
| (v) During hydro processing oxygen compounds may also cause rapid catalyst deactivation153 | ||
| Steam reforming | (i) Catalyst deactivation by coking and the formation of carbon deposits are major challenges | (i) The carbon deposition in fluidized bed is less severe than fixed bed164 |
| (ii) The steam reforming of bio-oil is still in an early stage of development mainly due the short lifetime of the catalysts with typical operating hours for acceptable rates being less than 100 h (ref. 156) | (ii) Catalyst deactivation by coking could be reduced by selectively reforming specific fractions of the bio-oil | |
| (iii) The hydrophobic fraction mainly composed of heavy oligomers can be separated by water extraction as a high value product |
3.8 Catalytic esterification
Catalytic esterification converts carboxylic acids into esters through the addition of an external alcohol, such as methanol or ethanol.165 Process is carried out to reduce the viscosity, acidity and oxygen content of the bio-oil which improves the stability of bio-oil. It also decreases the H2 consumption in subsequent hydro treating.165 Mineral acids, such as H2SO4, HCl, and H3PO4, are commonly used a catalyst for esterification.166 Corrosiveness of these mineral acids and difficulty in their recovery from products necessitate the need to develop a solid acid catalyst. Metal oxides, such as TiO2-, ZrO2-, and Fe2O3-containing mixed oxides, become highly acidic on modification with anions, such as SO42−, PO43−, etc. and these solid acid catalysts have been proposed for bio-oil upgrading.166 Other methods to improve the quality of bio-oil includes emulsification,167 distillation,168 solvent extraction169 and supercritical CO2 extraction.1703.9 Fermentation of sugar
Fermentation is an enzyme driven biochemical conversion of sugar into ethanol. The stoichiometric reaction for fermentations of glucose and xylulose to ethanol are expressed as under.| C6H12O6 → 2C2H5OH + 2CO2 |
| 3C5H10O5 → 5C2H5OH + 5CO2 |
The theoretical yield of ethanol is 0.51 g g−1 sugar for both glucose and xylulose. The complete process of biomass conversion into ethanol consists of four major steps: pre-treatment, hydrolysis, fermentation, and product upgradation to fuel quality. There are various routes for enzymatic hydrolysis and fermentation including (i) separate hydrolysis and fermentation (SHF) where enzymatic hydrolysis and fermentation are performed sequentially, (ii) simultaneous saccharification and fermentation (SSF) where enzymatic hydrolysis and fermentation are carried out simultaneously in the same vessel, simultaneous saccharification and cofermentation (SSCF) which involves two process steps: cellulase production followed by cellulose hydrolysis and the fermentation of both cellulose and hemicellulose and (iv) consolidated bioprocessing (CBP) where all bioconversion steps are completed in a single reactor.54 Ishola, et al.171 has presented a novel method for bioethanol production from lignocellulosic biomass involving simultaneous saccharification, filtration and fermentation (SSFF). SSFF was also examined for simultaneous glucose and xylose uptake.
Bai et al.172 has critically reviewed ethanol fermentation technologies from sugar and starch feedstocks. However, lignocellulosic biomass contains complex carbohydrates and the lack of industrially suitable microorganisms for converting biomass into fuel ethanol has traditionally been cited as a major technical roadblock in developing a bioethanol industry.173 Conversion up to 270 L per tonne of biomass has been achieved and NREL projects that by 2030, technology developments will enable yields of ethanol to approach 400 L per dry tonne.54 Yuan et al.174 demonstrated that industrially proven yeast strains such as ethanol red can convert high concentrations of mixed sugars to ethanol in about 30 h with yields comparable to engineered strains.
3.10 Synthesis gas fermentation
A significant portion of biomass is poorly degradable. The gasification of this non-degradable material to produce syngas followed by fermentation of synthetic gas to ethanol offer alternate option to convert poorly degradable biomass into ethanol and elimination the need of costly pre-treatment.175 Syngas-fermenting microorganisms use acetyl-CoA pathway to produce ethanol176 as shown in following equations.176| 6CO + 3H2O → C2H5OH + 4CO2, ΔH = −217.9 kJ mol−1 |
| 6CO2 + 6H2 → C2H5OH + 3H2O, ΔH = −97.3 kJ mol−1 |
Poor mass transfer properties of the gaseous substrates (mainly CO and H2) and low ethanol yield of gas phase fermentation are the biggest challenges preventing large scale adoption of syngas fermentation technology.175
3.11 Product development
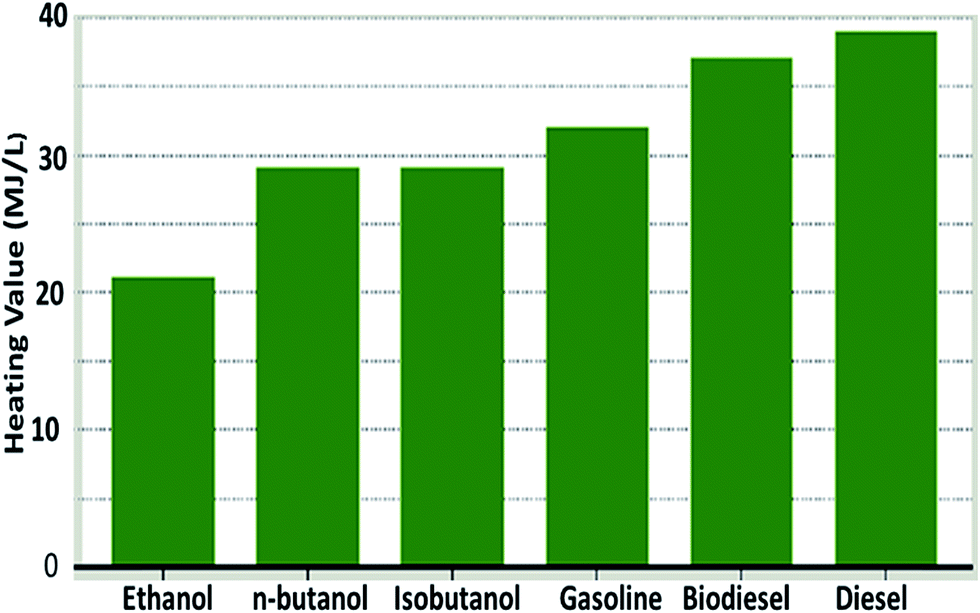 | ||
| Fig. 12 Comparison of heating value of selected fuels180 [reprinted from ref. 180 with permission from Macmillan Publishers Ltd]. | ||
ED20 (blend of diesel contains 15 per cent ethanol and 5 per cent emulsifiers) can be used in present day compression-ignition vehicles without any modification.181,182 Beside reducing the dependency on fossil fuel, oxygenated ED20 significantly reduces emissions of particulate matter (PM) and reduces toxic gases such as CO, SOx, and NOx from tailpipe emissions.183 The ED95 technology, which use 95% hydrous ethanol and 5% ignition improver with no diesel has also been developed.182 There are a number of additives, which improves the essential properties of fuel for proper operation of a diesel engine184 such as emulsifier increases the ethanol–diesel miscibility and cetane improver enhances the cetane number of ED.
| Iso-octane | Butanol | Ethanol | |
|---|---|---|---|
| Chemical formula | C8H18 | C4H10O | C2H6O |
| Low heating value (MJ kg−1) | 44.3 | 32 | 26.8 |
| Latent heat of vaporization (kJ L−1) | 213.1 | 474 | 725.4 |
| Density (kg m−3) | 692 | 813 | 794 |
| Oxygen (wt%) | 0 | 21.6 | 34.7 |
| RON | 100 | 113 | 111 |
Butanol it is much less anhydrous, which greatly reduces the risk of water contamination/absorption by the fuel.180 The energy content of iso-butanol (higher heating value of 36.0 MJ kg−1) is almost 80% that of ordinary gasoline (46.5 MJ kg−1), and is significantly higher than that of ethanol (29.7 MJ kg−1). Iso-butanol has a lower Reid vapor pressure than ethanol, making it more suitable as a gasoline fuel, also in colder climates. In addition, it has a higher density and a significantly lower oxygen content, which adds to its suitability as fuel in internal combustion engines. Iso-butanol can be used as a drop-in additive to gasoline, and also as a major gasoline component, without any modifications of ordinary internal combustion engines being required.186 Compared with unleaded gasoline, fuels blends containing different alcohols appear to have a lower carbon monoxide and hydrocarbon emissions and a higher fuel consumption rate and CO2 emissions.187
There are several conventional and new processes for conversion of biomass from municipal, agricultural and forest waste into bio-methanol such as pyrolysis, gasification, biosynthesis, electrolysis and photo electrochemical processes.188 In conventional methanol plant feedstock are converted into syngas through gasification.189 The main processes in a conventional methanol plant are gasification, gas cleaning, syngas conditioning and methanol synthesis and purification. The technologies used in the production of methanol from biomass are relatively well known since they are similar to the coal gasification technology, which has been applied for a long time. Usman and Wan Daud190 has summarized recent advances in methanol synthesis. However, making biomass gasification cost-competitive has proven difficult. The energy efficiency of methanol production from natural gas ranges about 60–70 per cent. However during methanol from biomass, the energy efficiency is around 50–60 per cent. Campo et al.191 showed a conversion efficiency of 54% in the production of bio methanol from biomass using a gasification process with an efficiency of 55%. The production cost of bio-methanol 1.5–4.0 times higher than production cost of natural gas-based methanol. New technologies such as photo electrochemical and electrolysis have potential for the production of bio-methanol at a lab scale but require further research prior to their use in large-scale.188
Both thermochemical and biochemical route can be used to produce H2 from biomass. The thermochemical route includes pyrolysis, gasification, steam gasification, steam reforming of bio-oils while biochemical route comprise of enzymatic decomposition of sugars. Thermochemical methods have some significance advantages over bio-chemical methods. Overall efficiency (thermal to H2) of thermochemical process is higher (52%) and production cost is lower when compared to bio-chemical process. The major disadvantage of thermochemical route is the decomposition of the biomass feed stock leading to char and tar formation. The cost of H2 production from supercritical water gasification of wet biomass was several times higher than the current price of H2 from steam methane reforming.194 Estimated cost comparison of H2 production by biomass gasification and natural gas steam reforming is shown in Fig. 13.
 | ||
| Fig. 13 Estimated cost comparison of hydrogen production by biomass gasification and natural gas steam reforming.195 | ||
Biological production of H2 is one of the alternative methods where processes can be operated at ambient temperatures and pressures, and are less energy intensive and more environmental friendly. Hydrolysis (acid or enzymatic) of starch/cellulose to highly concentrated sugar solution is the first step in fermentative H2 production from waste biomass followed by dark fermentation of resulting carbohydrates to volatile fatty acids, H2 and CO2 by acetogenic-anaerobic bacteria. A wide variety of heterotrophic bacteria have the ability to ferment carbohydrates under anaerobic conditions to produce H2 gas, volatile fatty acids and CO2.196
| C6H12O6 + 2H2O → 2CH3COOH + 4H2O + 2CO2, ΔG0 = −206 kJ |
Fermentative route of H2 production from biomass is a promising approach provided that the rate and the yields of H2 formation is improved to economically feasible levels and large scale operations is developed.196 Bio-hydrogen production processes are found to be more environmental friendly and less energy intensive as compared to thermo-chemical and electrochemical processes.197 Cortright and co-workers198 demonstrated that H2 can be produced from sugars and alcohols extracted from plant at temperatures near 225 °C in a single-reactor aqueous-phase reforming process using a platinum-based catalyst. Process breaks the glucose down into H2, CO2, and small amounts of CH4. The technique is even more efficient when methanol is used instead of glucose. Deluga and co-workers199 combine partial oxidation with steam reforming and the water–gas shift reaction to convert ethanol and ethanol–water mixtures directly into H2. Selectivity was ∼100% and >95% conversion by catalytic partial oxidation, with a residence time on rhodium–ceria catalysts of <10 milliseconds;
| C2H5OH + 2H2O + ½O2 → 2CO2 + 5H2, ΔHr = −50 kJ mol−1 |
In order to produce 99.99% pure H2, it needs to clean raw H2. Pressure swing adsorption or a membrane is used to purify H2. As the cost of biohydrogen is higher than the cost of natural gas based H2, carbon credit is required for it to be competitive with natural-gas-based H2.200
DME can use the existing LPG and natural gas infrastructures for transport and storage. DME having cetane number 55–60 has been promoted as a diesel substitute.203,204 DME has low auto-ignition temperature, which results in instantaneous vaporization when injected into the cylinder. Its high oxygen content (around 35% by mass) and the absence of C–C bonds in the molecular structure also facilitates in smooth combustion in diesel engine. The viscosity of DME is lower than that of diesel by a factor of about 20. As a result blending DME increases chances of leakage in pumps and fuel injectors. There are also lubrication issues with DME which necessitate addition of additives to increase the lubricity of DME.148 DME is in used as a transportation fuel in Canada in spite of its short history.201
Table 13 shows the comparison of key properties of DME and diesel fuel.204
| Property (unit/condition) | Unit | DME | Diesel fuel |
|---|---|---|---|
| Molar mass | g mol−1 | 46 | 170 |
| Carbon content | Mass% | 52.2 | 86 |
| Hydrogen content | Mass% | 13 | 14 |
| Oxygen content | Mass% | 34.8 | 0 |
| Carbon-to-hydrogen ratio | 0.337 | 0.516 | |
| Critical temperature | K | 400 | 708 |
| Critical pressure | MPa | 5.37 | 3.00a |
| Critical density | kg m−3 | 259 | — |
| Liquid density | kg m−3 | 667 | 831 |
| Relative gas density (air = 1) | 1.59 | — | |
| Cetane number | >55 | 40–50 | |
| Auto-ignition temperature | K | 508 | 523 |
| Stoichiometric air/fuel mass ratio | 9 | 14.6 | |
| Boiling point at 1 atm | K | 248.1 | 450–643 |
| Enthalpy of vapourization | kJ kg−1 | 467.13 | 300 |
| Lower heating value | MJ kg−1 | 27.6 | 42.5 |
| Gaseous specific heat capacity | kJ kg−1 K−1 | 2.99 | 1.7 |
| Ignition limits (Vol% in air) | Vol% in air | 3.4/18.6 | 0.6/6.5 |
| Modulus of elasticity | N m−2 | 6.37 × 108 | 1.49 × 109 |
| Kinematic viscosity of liquid | cSt | <0.1 | 3 |
| Surface tension (at 298 K) | N m−1 | 0.012 | 0.027 |
| Vapor pressure (at 298 K) | kPa | 530 | ≪10 |
DME production from biomass typically involve syngas generation through steam reforming of biomass which is converted to methanol followed by methanol dehydration to DME. Typical biomass to DME production facilities consist of (i) pre-treatment, (ii)gasification, (iii) gas cleaning-up, (iv) gas reforming to obtain appropriate H2:CO ratio (v) cleaning-up of syngas and finally (vi) DME synthesis.205 The conversion comprises the following set of reactions:Methanol synthesis;
| CO + 2H2 ↔ CH3OH, ΔHr = 90.3 kJ mol−1 |
| 2CH3OH ↔ CH3OCH3 + H2O, ΔHr = 23.4kJ mol−1 |
| H2O + CO ↔ H2 + CO2, ΔHr = 40.9 kJ mol−1 |
| 3H2 + 3CO ↔ CH3OCH3 + CO2, ΔHr = 258.6 kJ mol−1 |
DME can also be produced directly from syngas in a single step reaction over hybrid catalysts. Single step synthesis on a bifunctional catalyst have advantage of lower thermodynamic limitation compared to methanol synthesis route where due low concentration of methanol in the reaction medium shifts the thermodynamic equilibrium of methanol synthesis. Single step synthesis is carried out at higher temperature and lower pressure.206 CO2 incorporation in the feed is more feasible in single step route than in the synthesis of methanol, given that it requires lower pressure.206 In slurry reactor methanol synthesis, methanol dehydration to DME, and water–gas shift reactions all proceed concurrently.207 The process offers potential lower capital and operating costs. However, further research is needed for the development of novel catalysts which show better performance in terms of activity, selectivity and most importantly stability towards water.202 IEA reported that well-to-wheels energy efficiency of biomass gasification and conversion to DME was 56%.205
Biodiesel has significantly lower emissions than petroleum based diesel.211 Biodiesel is better than petroleum based diesel in terms of sulfur content, flash point, aromatic content and biodegradability. However due to higher viscosity and lower energy density pure biodiesel is not suitable to apply in the engines directly. Currently most vehicle and engine manufacturers have approved the usage of B5 biodiesel blend (5% biodiesel and 95% diesel by volume) in their engines with a large part of them have even raised the maximum limit up to B20.208
4. Availability and supply chain management of biomass feedstock
It is estimated that annually around 5.1 × 1012 kg agricultural residues and 501 × 109 kg forestry residues are produced globally.213 The biofuel industry is capital-intensive with large-scale economies and requires dependable year-round feedstock supply for continuous operation. Harvesting, treating, transporting, storing, and delivering large volumes of biomass feedstock, at a desired quality all-year-round to a biofuel plant requires careful logistical analysis prior to plant investment.214,215 Operational components of a biomass supply chain are shown in Fig. 14. Gold and Seuring216 has reviewed biomass supply chain and related issues. The parties involved in a biomass energy supply chain are: the supplier of biomass, transportation and distribution entities, energy production facility developers and operators, the government and utility firms who provide the incentives, and the end-users. Biomass energy supply chain differs from traditional supply chains in several ways. Among them are the seasonal availability of agricultural biomass, low energy density, demand variations due to uncertain energy production performance and the variability of biomass materials, which has implications for transport and storage. Thus, the main objectives of biomass supply chain management are to minimize costs, environmental impacts of the supply chain, and ensure continuous feedstock supply. | ||
| Fig. 14 Operational components of a biomass supply chain. | ||
Biomass feedstocks are highly dispersed and the distance biomass must be transported to a bioenergy facility is longer compared to the distance fossil fuels are transported to a facility with the same capacity. The transportation cost of biomass feedstock is also high compared to fossil fuel due to remote location of biomass resources. In addition to this biomass has low energy density compared to fossil fuels. These two characteristics of biomass make its delivery-cost high.200 In addition to being available at low cost, biomass feedstock must also be available on a very large scale to have a meaningful impact on energy and sustainability challenges. At present, there is no market for energy crops. Because of higher upfront costs for energy crops and the fact that it takes several years before energy crops can be harvested on an annual basis farmers need some type of purchase guarantees to convert land to energy crop plantations. Issues such as water use, fertilizer and pesticide application and runoff, and other sustainability matters have yet to be addressed, as well as the food versus energy issue.217
The information on the spatial availability of feedstock resources and its potential route flow is crucial to realistically model geographically explicitly biofuel production. Zhan et al.218 have designed a model based on geographic information system (GIS) for assessing the price policy to acquire feedstock. The study suggests that it is more suitable to pay a specific price unit weight to the feedstock supplier and then to pay the transport costs of feedstock than to pay a fixed price per tonne of feedstock sent by the supplier. Hess et al.219 has divided the feedstock portion of the cost three categories (i) grower payment which includes appropriate production costs and all other expenses related to the biomass value standing on the stump or in the field (ii) efficiency/capacity which involve overall supply system engineering and logistics costs, including equipment, labour, and consumables; and (iii) biomass quality cost adjustments based on composition, heat content, moisture, and particle size distribution. These categories are shown in following equation.219

Feedstock price is also influenced by competition from other crop alternatives and competing demands.220 Economic equilibrium theory states that commodity markets tend to seek a price at which supply meets demand, and this phenomenon balances perturbations of supply or demand that clears the market over time. Fig. 15 illustrates that the group of suppliers target feedstock supply capacity to meet the perceived demand at the supplier's absolute minimum selling price. Simulation model by Jeffers et al.221 shows that multiple policies targeting clean, domestically produced energy can create competition for biomass, and that this competition can effectively drive up prices for the biomass feedstocks and potentially exclude other sectors from the market. However, the feasibility planning depend on the profit potential, cost risk, and other factors affecting the decision on projects which help to avoid an overly optimistic expectation for all the involved parties.
 | ||
| Fig. 15 Causal loop diagram of a bioenergy feedstock market, with the dampening feedback loop in bold221 [reprinted from ref. 221 with permission from Elsevier]. | ||
Competitive quantities of biomass available for biofuel plant can be defined by a simple formula:
| Qj = qs − Qsoil − Qnc |
The gross residue amount Qi generated annually by the crop type i can be obtained by following equation222
| Qi = Yiηryri |
The residue to yield ratio (ηryri) indicates how much residue (mass) is generated per unit of crop products of crop type i. To estimate the actual potentially available biomass from the residues for biofuel production, it is necessary to establish the present utilization pattern of the residues and surplus availability factors (ηsuri). Considering all these factors, the annual theoretical available residue amount (field or process) Qthi for crop type i can be obtained as:
| Qthi = Yiηryriηsuri |
Sun et al., 2011 (ref. 223) established game model to analyse interactions among factors including unit procurement cost, unit transportation cost, basic price for crop residue collection. Let actual supply of agri-biomass Q is:
| Q = QA + QB, than Q ≤ kqsπrm2 |
Agricultural residue sources in close proximity are accorded higher preference and only when biomass sources in close proximity are exhausted more distant biomass are taken into account. Hence, the biomass collection area should be circular in order to minimize transportation costs. According to Sun et al.,223 the selling price of biomass (p) is function:


This proposition implies that the total transportation cost from the farmland to the central storage increases exponentially with increasing demands, making cheap agri-biomass costly. Statistics on the current usage of these residues is essential for calculating the surplus amounts of various biomass feedstock. A significant part of the agro-residues generated is consumed for fodder and manure at the source of generation. Other applications include their direct use as a cooking fuel, building construction and as raw material by the paper industry. Biofuel production has to compete with these industries to procure biomass. The total equilibrium order quantity of agri-biomass increases with both RA and RB until it reaches kqsrm2ct and ps directly affect Rj, j = A, B, the equilibrium supply and profits.
The collection and delivery method will vary with the type of residue, terrain, available machinery, location, soil, seasonal access etc. and the relative costs of collection. Assume that a biofuel plant is located at the centre of a circle of area A as shown in Fig. 16 with a radius of ro. So, the radius can be calculated for given annual feedstock processing capacity of biofuel plant (F) with help of following expression:

| F = FDO |
 | ||
| Fig. 16 Biomass collection area. | ||
The above equation is based on the assumption that the farmland is uniformly distributed. Collection costs depend on the spatial density, unit costs of recovery and capacity of the transportation units. Singh et al.224 has developed a mathematical model for collection and transporting the biomass from fields to biomass based power plant. According to model, the collection costs are the sum of total recovery costs for harvesting biomass and transport costs for moving the biomass from a loosely spread form to the transport unit. Let a local storage site be placed at location O in Fig. 16. Cellulosic biomass is collected and transported from a circular field surrounding the storage site is:


Total collection costs of biomass (Bc) in the field is:


Unit collection cost (Cc), defined as the ratio of total collection cost to the carrying capacity of transport unit (qc), will be:

Cost coefficient of road transport increases with an increase in collection area. The relationships of distance and density conspire to increase transportation costs. A common feature in optimized high volume long distance biomass supply chains is a pre-processing stage that includes particle size reduction and densification, but can also include pre-treatment of the biomass to facilitate both transport and downstream use.219
Current biomass harvesting and bailing machinery produce either rectangular (130–200 kg m−3) or round bales (60–100 kg m−3). The bulk density significantly influences the transportation and storage of biofuel feedstocks.225 Densification increase the bulk density which help large scale in storage, loading, and transportation of biomass. However biomass densification comes with added capital for machinery/energy cost and requires additional safety measures including dust control systems and spark detection and fire protection systems. Numerous vehicle movements will be required to transport the feedstock from central location to plant which necessities careful analysis of the infrastructure limitations, traffic congestion, and environmental impact.226 These factor influences in indirect costs. Rail transportation is cost-effective for medium to long overland transport distances (>100 km) involving stable and constant flow of goods.226 Hess et al.219 reported that the standardized uniform or advanced uniform supply logistic and equipment can increase efficiencies by commuting biomass feedstock to small sizes and improving bulk-handling efficiency and bulk density. Processes such as drying and torrefaction (i.e. reducing the moisture content with heating in the absence of oxygen), carbonization, pelletization, chopping, shredding, and grinding are some of pre-treatment approaches adopted by biomass energy industry. Supply logistics will become more important as development accelerates and competition for biomass feedstocks arises.
The transportation infrastructure required for shipment of liquid fuel from the refinery may also present a significant challenge. However, the distribution costs could be reduced through regional biofuel distribution.227 Bio-ethanol and bio-diesel are banned by most U.S. pipeline operators due to their polarity and other corrosion, contamination issues.228 Therefore, the current supply chains for ethanol and bio-diesel require dedicated fuel distribution and blending system. The most common practice is to transport bio-ethanol or bio-diesel from the biofuel production facility to distribution terminals by train, barge, or truck.228
5. Commercialization of biomass-to-liquid fuels processes and biofuel support measures
5.1 Biofuels support policies
The support for biofuels generally includes policies that mandate certain levels of use of biofuels, policies that offer subsidies or tax credits for biofuel production their use, and research initiatives to fill the knowledge gap. More than 50 countries have adopted blending targets or mandates while several other countries have announced biofuel quotas for future years. European Union Renewable Energy Directive (2009/28/EC) set a mandatory target of a 10 per cent renewable transport fuels ((RTF) for 2020.229 US “20 in 10” initiative call for the replacing 15% of the projected gasoline consumption by renewable fuels by 2017.230 US ethanol use is projected to increase from 55.2 × 109 L in the 2013–2014 to around 70 × 109 L in 2022–2023. Brazil has increased the ethanol blending rate from 25 per cent to 27 per cent3. The present ethanol requirements were 1.330 × 109 L for 5 per cent blending. IEA technology roadmap-biofuels for transport envisions that by 2050 biofuels will provide 27 per cent of world transport fuel.11 Advanced biofuels are assumed to be commercially available at scale from 2020 onwards reaching more than 10 per cent of road transport fuel demand by 2040.35.2 Volume of biofuels production
The low cost of feedstock, simple and integrated conversion process, and multi-products production contribute to the competitiveness of bioethanol production from lignocellulosic biomass.231 Brown and Brown has provided complete review of the current direction of cellulosic biofuel commercialization in US.232 In 2014, US ethanol plants produced 54.13 × 109 L of ethanol which was 58 per cent of world ethanol production. Brazil produced roughly 23.47 × 109 L ethanol and was responsible for about 25% of world production, while the European Union followed with 6 per cent. Fig. 17233 shows the volume of world ethanol, biodiesel and hydro treated vegetable oil production from 2004 to 2014 while Fig. 18234 shows the volume of ethanol produced by major ethanol producing countries. Fig. 19 shows US ethanol exports by destination in 2014.233 | ||
| Fig. 17 World ethanol, biodiesel and hydro treated vegetable oil production from 2004 to 2014.233 | ||
 | ||
| Fig. 18 World fuel ethanol production between 1990–2014.234 | ||
 | ||
| Fig. 19 US ethanol exports by destination in 2014.233 | ||
World biodiesel production in 2014 was 54.13 × 109 L, largely from fatty acid methyl ester (FAME) and a small quantity from hydrotreated vegetable oil (HVO). World biodiesel production is largely based on vegetable oils, mostly from rapeseed and soybeans with smaller shares from palm jatropha and coconut.235 Small amount of biodiesel is also produced from industrial by-products such as used cooking oils and animal fat. In the United States, the largest biodiesel production facility is owned by RBF Port Neches LLC while Diester Industrie of France is largest biodiesel producer in EU with production capacity of 2.8 × 109 L.
5.3 Refinery's perspective on co-processing with crude oil
Crude bio-oil forms an alternative source of feedstock for the refineries. Bio-oils co-processing together with crude oil feedstocks is technically feasible and an attractive option236 with advantage of economy of scale.237 However such option increases refinery processing expanses due to a decrease in quality of combined stream oil.237,238 Pinho et al.236 directly co-processed bio-oil without any type of hydrodeoxygenation (HDO) with a regular gasoil fluid catalytic cracking (FCC) feed. For 10% bio-oil in the feed, 2% renewable carbon was obtained in the total liquid product.236 A high amount of phenolic compounds was detected in the naphtha produced by the FCC. Directly upgrade bio-oil in the FCC results in production of significant amounts of char, coke and water as the main products.239 Bio-oil immiscibility with hydrocarbons has also been singled out as an impediment to bio-oil direct introduction in the FCC processes.237 Some authors suggest a previous stabilization HDO step for the bio-oils prior to feeding them into the FCC. Special care is required during the selection of materials used for bio-oil storage and feed lines in the FCC unit to deal with the high acidity of the bio-oil.236 The alkaline metals present in the bio-oil destroy the zeolite in the FCC catalyst which result in higher catalyst to ensure FCC equilibrium catalyst activity.2365.4 Cost of producing advanced biofuels
Establishing next generation bioethanol facilities require heavier investment than first generation facility of similar capacity.240 Based upon the fuel conversion efficiency which is explained by the amount of sugars available after biomass pre-treatment ethanol produced by fermentation is unable to match the thermo-chemical conversion. The ethanol production from lignocellulosic ethanol seldom reaches higher than 4% (w/w) which requires energy intense distillation for upgradation to fuel quality.241 This results in overall low output. According techno-economic comparison analysis by Anex et al.38 stand-alone biomass-to-liquid fuel plants are expected to produce fuels with a product value in the range of $ 0.5–1.50 per L gasoline equivalent, with fast pyrolysis being the lowest, and bio-chemical conversion the highest.38 With ongoing research most of the technologies discussed here present a good potential to overcome technical bottlenecks, however they will not likely be cost-competitive with fossil fuels unless a climate policy is in place.5.5 Bio-refinery facilities
In United States around 213 bio refinery facilities are producing a range of co-products with ethanol while another 100 were expanding or under construction in 2014. The advanced biofuel production facilities that came on line in 2014 included three new bio refineries using cellulosic plant material in the United States: POET-DSM, DuPont, and Abengoa. DuPont is developing Nevada, Iowa cellulosic ethanol facility to produce 115 × 106 L of fuel-grade ethanol in its stover-to-ethanol bio refinery consuming 700 × 103 corn stover bales per year. DuPont also announced to develop a plant to produce cellulosic ethanol in Siping City, using residues from corn production in Jilin Province of China.The Beta Renewables commercial scale cellulosic ethanol plant at Crescentino has a production capacity of 75 × 106 L cellulosic ethanol annually using non-food biomass such as agricultural residues. In Brazil, the three commercial second-generation biofuel projects started operation include GranBio commercial cellulosic ethanol plant, Raizen/Iogens plant, and Solazyme-Bunge plant. GranBio has initial production capacity of 82 × 106 L of ethanol per year using the agricultural waste – straw and bagasse. The major companies in the world biofuel industry incudes US producer Archer Daniels Midland (ADM), which owns the five largest ethanol plants in the world with total production capacity of 5 × 109 L per y, Denmark's Novozymes who provides enzymes for about 60% of global corn ethanol production; DuPont which is the second largest supplier of industrial enzymes. The other players are Odebrecht Agroindustrial (Brazil), Abengoa Bioenergy (Spain), and Henan Tianguan Group (China).
6. Conclusion and future outlook
The production of liquid transportation fuels from lignocellulosic biomass is a worldwide strategy for the reduction of environmental pollution of fossil fuel combustions. Liquid biofuels are compatible with the existing infrastructure for distribution and delivery of transport fuel. Liquid biofuel produced from non-food feedstock is a viable substitution for petroleum-derived fuels in the transport sector. Production of liquid biofuel from local resources also enhance energy security and mitigate global warming without taking food away from a hungry planet. Recently the production of liquid biofuels for transport sector has exhibited the most rapid growth, fostered by government support. The incentives for use of renewable resources to replace fossil fuels are also motivating extensive research to derive liquid fuels from biomass. For transition to next-generation liquid biofuels is necessary to increase their production and to reduce use of fossil fuel. The flexible energy system, which uses both biomass and fossil fuels in combination, could be the backbone for a low risk, low cost and low carbon emission energy supply system.Both thermo-chemical and bio-chemical processes produces liquid fuels suitable for use as transportation fuels. Thermo-chemical route produces liquid fuels more suitable for diesel cycle engines while bio-chemical based alcohols are suitable for use in spark ignition engines. At present neither the biochemical nor thermochemical platforms have clear advantages in capital costs nor operating costs for production of advanced biofuels. However thermochemical process converts the biomass in only a few seconds whereas traditional enzymatic approaches takes few to hours or days for conversion. An approach to improve the economic viability and sustainability of the utilization of BTL fuel is the combined production of more than one product. Little information is available on integration of biochemical and thermochemical route. This can be further explored to overcome individual problems being facing by both routes. The development of more robust biological processing options could allow for straight forward production of alcohols and specialty chemicals, both of which could add flexibility and value to an integrated bio-refinery. Process integration research focused on modeling unit operations adequately evaluated on the bases of the entire pathway can help in optimization of performance of individual operation as well as whole pathway to improve the performance and reduce the production cost. Other constraints in commercial scale liquid biofuel production included high production costs, unstable feedstock supplies, logistical challenges, and policy uncertainty. For next-generation liquid biofuels the competitiveness will increase in mid- to long-term due to economies of scale and learning curve effects. The sustainable trade in liquid biofuels also increase the important for the supply of biomass and also trigger investments and mobilize biomass potentials. To achieve lower production costs, a consistent and sustainable supply of cheap raw materials is essential. The effective solution need a guaranteed supply of feedstock as well as solutions for logistic and economic issues. Ultimately, the world hope to see a holistic approach for cleaner production of liquid biofuel utilizing the local resources.
Acknowledgements
This work is financially supported by Mehran University of Engineering and Technology (MUET), Jamshoro, Pakistan and National Natural Science Foundation of China (21176021, 21276020), and the Fundamental Research Funds for the Central Universities (YS1401), and the Ministry of Science and Technology (863 Program, 2012AA101803).References
- D. Streimikiene, T. Baležentis and L. Baležentienė, Renewable Sustainable Energy Rev., 2013, 20, 611–618 CrossRef CAS.
- C. Linton, S. Grant-Muller and W. F. Gale, Transport Rev., 2015, 35, 533–553 CrossRef.
- IEA, Energy Climateand Change: World Energy Outlook Special Report, International Energy Agency (IEA), Paris, France, 2015 Search PubMed.
- S. K. Ribeiro, S. Kobayashi, M. Beuthe, J. Gasca, D. Greene, D. S. Lee, Y. Muromachi, P. J. Newton, S. Plotkin, D. Sperling, R. Wit and P. J. Zhou, Transport and its infrastructure, Cambridge, United Kingdom and New York, NY, USA, 2007 Search PubMed.
- R. C. Pietzcker, T. Longden, W. Chen, S. Fu, E. Kriegler, P. Kyle and G. Luderer, Energy, 2014, 64, 95–108 CrossRef.
- D. Sperling and S. Yeh, Transport Pol., 2010, 17, 47–49 CrossRef.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 CAS.
- K. Sanderson, Nature, 2011, 474, S12–S14 CrossRef CAS PubMed.
- A. H. Sebayang, H. H. Masjuki, H. C. Ong, S. Dharma, A. S. Silitonga, T. M. I. Mahlia and H. B. Aditiya, RSC Adv., 2016, 6, 14964–14992 RSC.
- S. Lee and Y. T. Shah, Biofuels and Bioenergy: Processes and Technologies, Taylor & Francis, 2012 Search PubMed.
- IEA, Technology Roadmap-Biofuels for Transport, International Energy Agency (IEA), Paris Cedex 15, France, 2011 Search PubMed.
- A. Demirbas, Appl. Energy, 2009, 86, S108–S117 CrossRef CAS.
- J. R. Regalbuto, Science, 2009, 325, 822–824 CrossRef PubMed.
- H. Long, X. Li, H. Wang and J. Jia, Renewable Sustainable Energy Rev., 2013, 26, 344–352 CrossRef.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- M. S. Mettler, D. G. Vlachos and P. J. Dauenhauer, Energy Environ. Sci., 2012, 5, 7797–7809 CAS.
- M. J. A. Tijmensen, A. P. C. Faaij, C. N. Hamelinck and M. R. M. van Hardeveld, Biomass Bioenergy, 2002, 23, 129–152 CrossRef CAS.
- A. Pandey, T. Bhaskar, M. Stöcker and R. K. Sukumaran, in Recent Advances in Thermo-Chemical Conversion of Biomass, ed. A. Pandey, T. Bhaskar, M. Stöcker and R. K. Sukumaran, Elsevier, Boston, 2015, pp. xi–xii Search PubMed.
- S. Zinoviev, F. Müller-Langer, P. Das, N. Bertero, P. Fornasiero, M. Kaltschmitt, G. Centi and S. Miertus, ChemSusChem, 2010, 3, 1106–1133 CrossRef CAS PubMed.
- A. W. Bhutto, A. A. Bazmi and G. Zahedi, Prog. Energy Combust. Sci., 2013, 39, 189–214 CrossRef CAS.
- H. A. Baloch, T. Yang, R. Li, S. Nizamuddin, X. Kai and A. W. Bhutto, Clean Technol. Environ. Policy, 2016 DOI:10.1007/s10098-016-1092-4.
- P. L. Spath and D. C. Dayton, Preliminary Screening – Technical and Economic Assessment of Synthesis Gas to Fuels and Chemicals with Emphasis on the Potential for Biomass-Derived Syngas, National Renewable Energy Laboratory, Cole Boulevard Golden, Colorado, 2003 Search PubMed.
- S. Heidenreich and P. U. Foscolo, Prog. Energy Combust. Sci., 2015, 46, 72–95 CrossRef.
- D. L. Klass, Biomass for Renewable Energy, Fuels and Chemicals, Academic Press, San Diego, 1998 Search PubMed.
- S. Bhaduri, F. Contino, H. Jeanmart and E. Breuer, Energy, 2015, 87, 289–302 CrossRef CAS.
- E. Kırtay, Energy Convers. Manage., 2011, 52, 1778–1789 CrossRef.
- C. F. Palma, Appl. Energy, 2013, 111, 129–141 CrossRef.
- P. De Filippis, M. Scarsella, B. de Caprariis and R. Uccellari, Energy Procedia, 2015, 75, 240–245 CrossRef CAS.
- J. Han and H. Kim, Renewable Sustainable Energy Rev., 2008, 12, 397–416 CrossRef CAS.
- P. J. Woolcock and R. C. Brown, Biomass Bioenergy, 2013, 52, 54–84 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- L. Faba, E. Díaz and S. Ordóñez, Renewable Sustainable Energy Rev., 2015, 51, 273–287 CrossRef CAS.
- A. Anca-Couce, Prog. Energy Combust. Sci., 2016, 53, 41–79 CrossRef.
- P. C. Badger and P. Fransham, Biomass Bioenergy, 2006, 30, 321–325 CrossRef CAS.
- P. McKendry, Bioresour. Technol., 2002, 83, 47–54 CrossRef CAS PubMed.
- R. H. Venderbosch and W. Prins, Biofuels, Bioprod. Biorefin., 2010, 4, 178–208 CrossRef CAS.
- R. Agrawal and N. R. Singh, AIChE J., 2009, 55, 1898–1905 CrossRef CAS.
- R. P. Anex, A. Aden, F. K. Kazi, J. Fortman, R. M. Swanson, M. M. Wright, J. A. Satrio, R. C. Brown, D. E. Daugaard, A. Platon, G. Kothandaraman, D. D. Hsu and A. Dutta, Fuel, 2010, 89, S29–S35 CrossRef CAS.
- C. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Chem. Soc. Rev., 2014, 43, 7594–7623 RSC.
- A. V. Bridgwater, Environ. Prog. Sustainable Energy, 2012, 31, 261–268 CrossRef CAS.
- A. V. Bridgwater, D. Meier and D. Radlein, Org. Geochem., 1999, 30, 1479–1493 CrossRef CAS.
- P. K. Kanaujia, Y. K. Sharma, M. O. Garg, D. Tripathi and R. Singh, J. Anal. Appl. Pyrolysis, 2014, 105, 55–74 CrossRef CAS.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 CAS.
- Z. Yang, A. Kumar and R. L. Huhnke, Renewable Sustainable Energy Rev., 2015, 50, 859–870 CrossRef CAS.
- C. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Chem. Soc. Rev., 2014, 43, 7594–7623 RSC.
- G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184–7201 CrossRef CAS PubMed.
- G. Yildiz, F. Ronsse, R. v. Duren and W. Prins, Renewable Sustainable Energy Rev., 2016, 57, 1596–1610 CrossRef CAS.
- R. Singh, A. Prakash, B. Balagurumurthy and T. Bhaskar, in Recent Advances in Thermo-Chemical Conversion of Biomass, Elsevier, Boston, 2015, pp. 269–291, DOI:10.1016/b978-0-444-63289-0.00010-7.
- Y. Xue, H. Chen, W. Zhao, C. Yang, P. Ma and S. Han, Int. J. Energy Res., 2016 DOI:10.1002/er.3473.
- Y. Qian, C. Zuo, J. Tan and J. He, Energy, 2007, 32, 196–202 CrossRef CAS.
- Z. Liu and F.-S. Zhang, Energy Convers. Manage., 2008, 49, 3498–3504 CrossRef CAS.
- S. Nizamuddin, N. Jayakumar, J. Sahu, P. Ganesan, A. Bhutto and N. Mubarak, Korean J. Chem. Eng., 2015, 32, 1789–1797 CrossRef CAS.
- H.-j. Huang and X.-z. Yuan, Prog. Energy Combust. Sci., 2015, 49, 59–80 CrossRef.
- A. W. Bhutto, K. Qureshi, K. Harijan, G. Zahedi and A. Bahadori, RSC Adv., 2014, 4, 3392–3412 RSC.
- G. Banerjee, J. Scott-Craig and J. Walton, BioEnergy Res., 2010, 3, 82–92 CrossRef.
- B. Hahn-Hägerdal, M. Galbe, M. F. Gorwa-Grauslund, G. Lidén and G. Zacchi, Trends Biotechnol., 2006, 24, 549–556 CrossRef PubMed.
- L. R. Lynd, W. H. v. Zyl, J. E. McBride and M. Laser, Curr. Opin. Biotechnol., 2005, 16, 577–583 CrossRef CAS PubMed.
- P. R. Stuart and M. M. El-Halwagi, Integrated Biorefineries: Design, Analysis, and Optimization, CRC Press, 2012 Search PubMed.
- M. Germec, K. Tarhan, E. Yatmaz, N. Tetik, M. Karhan, A. Demirci and I. Turhan, Biotechnol. Prog., 2016 DOI:10.1002/btpr.2225.
- A. Aden and T. Foust, Cellulose, 2009, 16, 535–545 CrossRef CAS.
- L. R. Lynd, M. S. Laser, D. Bransby, B. E. Dale, B. Davison, R. Hamilton, M. Himmel, M. Keller, J. D. McMillan, J. Sheehan and C. E. Wyman, Nat. Biotechnol., 2008, 26, 169–172 CrossRef CAS PubMed.
- A. Dutta, N. Dowe, K. N. Ibsen, D. J. Schell and A. Aden, Biotechnol. Prog., 2010, 26, 64–72 CAS.
- D. Klein-Marcuschamer, P. Oleskowicz-Popiel, B. A. Simmons and H. W. Blanch, Biomass Bioenergy, 2010, 34, 1914–1921 CrossRef CAS.
- F. K. Kazi, J. A. Fortman, R. P. Anex, D. D. Hsu, A. Aden, A. Dutta and G. Kothandaraman, Fuel, 2010, 89, S20–S28 CrossRef CAS.
- M. Jin, C. Gunawan, N. Uppugundla, V. Balan and B. E. Dale, Energy Environ. Sci., 2012, 5, 7168–7175 CAS.
- M. C. Coimbra, A. Duque, F. Saéz, P. Manzanares, C. H. Garcia-Cruz and M. Ballesteros, Renewable Energy, 2016, 86, 1060–1068 CrossRef CAS.
- C. Álvarez, F. M. Reyes-Sosa and B. Díez, Microb. Biotechnol., 2016, 9, 149–156 CrossRef PubMed.
- M. Knauf and M. Moniruzzaman, Int. Sugar J., 2004, 106, 147–150 CAS.
- S. Kumar, P. Dheeran, S. P. Singh, I. M. Mishra and D. K. Adhikari, Renewable Energy, 2015, 83, 850–858 CrossRef CAS.
- T. Tsoutsos and D. Bethanis, Energies, 2011, 4, 1601 CrossRef CAS.
- P. Lenihan, A. Orozco, E. O'Neill, M. N. M. Ahmad, D. W. Rooney and G. M. Walker, Chem. Eng. J., 2010, 156, 395–403 CrossRef CAS.
- L. Kupiainen, PhD, University of Oulu, 2012.
- A. DemİRbaŞ, Energy Sources, 2005, 27, 327–337 CrossRef.
- A. K. Chandel, E. Chan, R. Rudravaram, M. L. Narasu, L. V. Rao and P. Ravindra, Biotechnol. Mol. Biol. Rev., 2007, 2, 14–32 Search PubMed.
- A. Shahbazi and B. Zhang, in Bioalcohol Production, Woodhead Publishing, 2010, pp. 143–158, DOI:10.1533/9781845699611.2.143.
- G. Hilpmann, N. Becher, F. A. Pahner, B. Kusema, P. Mäki-Arvela, R. Lange, D. Y. Murzin and T. Salmi, Catal. Today, 2016, 259(2), 376–380 CrossRef CAS.
- P. Walden, Bull. Russ. Acad. Sci., 1914, 405–422, DOI:12856962.
- F. H. Hurley and T. P. WIer, J. Electrochem. Soc., 1951, 98, 207–212 CrossRef CAS.
- S. E. Fry and N. J. Pienta, J. Am. Chem. Soc., 1985, 107, 6399–6400 CrossRef CAS.
- J. A. Boon, J. A. Levisky, J. L. Pflug and J. S. Wilkes, J. Org. Chem., 1986, 51, 480–483 CrossRef CAS.
- R. Abro, A. A. Abdeltawab, S. S. Al-Deyab, G. Yu, A. B. Qazi, S. Gao and X. Chen, RSC Adv., 2014, 4(67), 35302–35317 RSC.
- A. Brandt, J. Grasvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- T. Heinze and T. Liebert, Prog. Polym. Sci., 2001, 26, 1689–1762 CrossRef CAS.
- T. Liebert and T. Heinze, BioResources, 2008, 3, 576–601 Search PubMed.
- S. Barthel and T. Heinze, Green Chem., 2006, 8, 301–306 RSC.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- J. Wu, J. Zhang, H. Zhang, J. He, Q. Ren and M. Guo, Biomacromolecules, 2004, 5, 266–268 CrossRef CAS PubMed.
- H. Zhang, J. Wu, J. Zhang and J. He, Macromolecules, 2005, 38, 8272–8277 CrossRef CAS.
- C. Graenacher, US Pat. 1943176, 1934.
- I. Kilpeläinen, H. Xie, A. King, M. Granstrom, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142–9148 CrossRef PubMed.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- P. Mäki-Arvela, I. Anugwom, P. Virtanen, R. Sjöholm and J. P. Mikkola, Ind. Crops Prod., 2010, 32, 175–201 CrossRef.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Energy Fuels, 2010, 24, 737–745 CrossRef CAS.
- A. George, A. Brandt, K. Tran, S. M. S. N. S. Zahari, D. Klein-Marcuschamer, N. Sun, N. Sathitsuksanoh, J. Shi, V. Stavila, R. Parthasarathi, S. Singh, B. M. Holmes, T. Welton, B. A. Simmons and J. P. Hallett, Green Chem., 2015, 17, 1728–1734 RSC.
- N. Muhammad, Y. A. Elsheikh, M. I. A. Mutalib, A. A. Bazmi, R. A. Khan, H. Khan, S. Rafiq, Z. Man and I. khan, J. Ind. Eng. Chem., 2015, 21, 1–10 CrossRef CAS.
- C. Li, Q. Wang and Z. K. Zhao, Green Chem., 2008, 10, 177–182 RSC.
- C. Z. Li and Z. K. B. Zhao, Adv. Synth. Catal., 2007, 349, 1847–1850 CrossRef CAS.
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47, 8047–8050 CrossRef CAS PubMed.
- M. B. Turner, S. K. Spear, J. G. Huddleston, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 443–447 RSC.
- A. M. Socha, R. Parthasarathi, J. Shi, S. Pattathil, D. Whyte, M. Bergeron, A. George, K. Tran, V. Stavila, S. Venkatachalam, M. G. Hahn, B. A. Simmons and S. Singh, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3587–E3595 CrossRef CAS PubMed.
- D. Klein-Marcuschamer, B. A. Simmons and H. W. Blanch, Biofuels, Bioprod. Biorefin., 2011, 5, 562–569 CrossRef CAS.
- F. Xu, J. Sun, N. V. S. N. M. Konda, J. Shi, T. Dutta, C. D. Scown, B. A. Simmons and S. Singh, Energy Environ. Sci., 2016, 9, 1042–1049 CAS.
- J. Rodrigues, I. Miranda, J. Gominho, M. Vasconcelos, G. Barradas, H. Pereira, F. Bianchi-de-Aguiar and S. Ferreira-Dias, Ind. Crops Prod., 2016, 83, 614–619 CrossRef CAS.
- R. C. Pradhan, S. Mishra, S. N. Naik, N. Bhatnagar and V. K. Vijay, Biosystems Engineering, 2011, 109, 158–166 CrossRef.
- S. Karaj and J. Müller, Ind. Crops Prod., 2011, 34, 1010–1016 CrossRef.
- R. N. Patel, S. Bandyopadhyay and A. Ganesh, Energy Convers. Manage., 2011, 52, 652–657 CrossRef CAS.
- M. J. H. Akanda, M. Z. I. Sarker, S. Ferdosh, M. Y. A. Manap, N. N. N. Ab Rahman and M. O. Ab Kadir, Molecules, 2012, 17, 1764–1794 CrossRef CAS PubMed.
- C. P. Passos, M. A. Coimbra, F. A. Da Silva and C. M. Silva, Chem. Eng. Res. Des., 2011, 89, 1118–1125 CrossRef CAS.
- R. Vardanega, D. T. Santos and M. A. A. Meireles, Pharmacogn. Rev., 2014, 8, 88–95 CrossRef PubMed.
- M. M. R. de Melo, A. J. D. Silvestre and C. M. Silva, J. Supercrit. Fluids, 2014, 92, 115–176 CrossRef CAS.
- S. P. Singh and D. Singh, Renewable Sustainable Energy Rev., 2010, 14, 200–216 CrossRef CAS.
- M. Kouzu and J.-s. Hidaka, Fuel, 2012, 93, 1–12 CrossRef CAS.
- S. S. Ail and S. Dasappa, Renewable Sustainable Energy Rev., 2016, 58, 267–286 CrossRef CAS.
- D. Y. C. Leung, X. Wu and M. K. H. Leung, Appl. Energy, 2010, 87, 1083–1095 CrossRef CAS.
- J. M. Encinar, A. Pardal and N. Sánchez, Fuel, 2016, 166, 51–58 CrossRef CAS.
- F. H. Kasim and A. P. Harvey, Chem. Eng. J., 2011, 171, 1373–1378 CrossRef CAS.
- S. R. Dasari, V. B. Borugadda and V. V. Goud, Process Saf. Environ. Prot., 2016, 100, 252–263 CrossRef CAS.
- S. H. Mushrif, V. Vasudevan, C. B. Krishnamurthy and B. Venkatesh, Chem. Eng. Sci., 2015, 121, 217–235 CrossRef CAS.
- I. Narváez, A. Orío, M. P. Aznar and J. Corella, Ind. Eng. Chem. Res., 1996, 35, 2110–2120 CrossRef.
- Y. Shen, Renewable Sustainable Energy Rev., 2015, 43, 281–295 CrossRef CAS.
- A. Sharma, V. Pareek and D. Zhang, Renewable Sustainable Energy Rev., 2015, 50, 1081–1096 CrossRef CAS.
- A. H. Zacher, M. V. Olarte, D. M. Santosa, D. C. Elliott and S. B. Jones, Green Chem., 2014, 16, 491–515 RSC.
- E. Kantarelis, doctoral dissertation, KTH-Royal Institute of Technology, 2014.
- D. Chen, Z. Zheng, K. Fu, Z. Zeng, J. Wang and M. Lu, Fuel, 2015, 159, 27–32 CrossRef CAS.
- O. Das and A. K. Sarmah, Sci. Total Environ., 2015, 538, 145–151 CrossRef CAS PubMed.
- D. C. Elliott, P. Biller, A. B. Ross, A. J. Schmidt and S. B. Jones, Bioresour. Technol., 2015, 178, 147–156 CrossRef CAS PubMed.
- S. S. Toor, L. Rosendahl and A. Rudolf, Energy, 2011, 36, 2328–2342 CrossRef CAS.
- R. Shakya, J. Whelen, S. Adhikari, R. Mahadevan and S. Neupane, Algal Res., 2015, 12, 80–90 CrossRef.
- Y. Guo, T. Yeh, W. Song, D. Xu and S. Wang, Renewable Sustainable Energy Rev., 2015, 48, 776–790 CrossRef CAS.
- S. Karagöz, T. Bhaskar, A. Muto, Y. Sakata, T. Oshiki and T. Kishimoto, Chem. Eng. J., 2005, 108, 127–137 CrossRef.
- J. B. Binder and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4516–4521 CrossRef CAS PubMed.
- S. J. B. Duff and W. D. Murray, Bioresour. Technol., 1996, 55, 1–33 CrossRef CAS.
- Y. Sun and J. Cheng, Bioresour. Technol., 2002, 83, 1–11 CrossRef CAS PubMed.
- S. T. Moe, K. K. Janga, T. Hertzberg, M.-B. Hägg, K. Øyaas and N. Dyrset, Energy Procedia, 2012, 20, 50–58 CrossRef CAS.
- P. C. Badger, in Trends in new crops and new uses, ed. J. Janick and A. Whipkey, ASHS Press, Alexandria, VA, 2002, pp. 17–21 Search PubMed.
- T. W. Jeffries and Y.-S. Jin, in Advances in Applied Microbiology, Academic Press, 2000, vol. 47, pp. 221–268 Search PubMed.
- A. Demirbas, Prog. EnergyCombust. Sci., 2007, 33, 1–18 CrossRef CAS.
- A. Demirbaş, Int. J. Green Energy, 2004, 1, 79–87 CrossRef.
- S. M. Sen, J. B. Binder, R. T. Raines and C. T. Maravelias, Biofuels, Bioprod. Biorefin., 2012, 6, 444–452 CrossRef CAS.
- J. Singh and P. C. Bargale, J. Food Eng., 2000, 43, 75–82 CrossRef.
- S. Al-Zuhair, Biofuels, Bioprod. Biorefin., 2007, 1, 57–66 CrossRef CAS.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- R. A. Friedel and R. B. Anderson, J. Am. Chem. Soc., 1950, 72, 1212–1215 CrossRef CAS.
- E. de Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758–2781 RSC.
- M. M. Wright and R. C. Brown, Biofuels, Bioprod. Biorefin., 2007, 1, 49–56 CrossRef CAS.
- D. Unruh, K. Pabst and G. Schaub, Energy Fuels, 2010, 24, 2634–2641 CrossRef CAS.
- A. Martínez and C. López, Appl. Catal., A, 2005, 294, 251–259 CrossRef.
- T. A. Semelsberger, R. L. Borup and H. L. Greene, J. Power Sources, 2006, 156, 497–511 CrossRef CAS.
- R. Luque, A. R. de la Osa, J. M. Campelo, A. A. Romero, J. L. Valverde and P. Sanchez, Energy Environ. Sci., 2012, 5, 5186–5202 CAS.
- H. Boerrigter, H. den-Uil and H. P. Calis, presented in part at the Pyrolysis and Gasification of Biomass and Waste-Expert Meeting, Strasbourge France, September 30, 2002 Search PubMed.
- Q. Zhang, J. Chang, T. Wang and Y. Xu, Energy Convers. Manage., 2007, 48, 87–92 CrossRef CAS.
- L. Zhang, R. Liu, R. Yin and Y. Mei, Renewable Sustainable Energy Rev., 2013, 24, 66–72 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- I. Gandarias, V. L. Barrio, J. Requies, P. L. Arias, J. F. Cambra and M. B. Güemez, Int. J. Hydrogen Energy, 2008, 33, 3485–3488 CrossRef CAS.
- M. Patel and A. Kumar, Renewable Sustainable Energy Rev., 2016, 58, 1293–1307 CrossRef CAS.
- R. Trane, S. Dahl, M. S. Skjøth-Rasmussen and A. D. Jensen, Int. J. Hydrogen Energy, 2012, 37, 6447–6472 CrossRef CAS.
- K. A. Resende, C. N. Ávila-Neto, R. C. Rabelo-Neto, F. B. Noronha and C. E. Hori, Renewable Energy, 2015, 80, 166–176 CrossRef CAS.
- X. Zhang, J. Long, W. Kong, Q. Zhang, L. Chen, T. Wang, L. Ma and Y. Li, Energy Fuels, 2014, 28, 2562–2570 CrossRef CAS.
- M. Asadieraghi, W. M. Ashri Wan Daud and H. F. Abbas, RSC Adv., 2015, 5, 22234–22255 RSC.
- J. Peng, P. Chen, H. Lou and X. Zheng, Bioresour. Technol., 2009, 100, 3415–3418 CrossRef CAS PubMed.
- R. S. Weber, M. V. Olarte and H. Wang, Energy Fuels, 2015, 29, 273–277 CrossRef CAS.
- G. Haarlemmer and T. Bensabath, Comput. Chem. Eng., 2016, 84, 281–289 CrossRef CAS.
- X. Guo, Y. Zheng, B. Zhang and J. Chen, Biomass Bioenergy, 2009, 33, 1469–1473 CrossRef CAS.
- P. Lan, Q. Xu, M. Zhou, L. Lan, S. Zhang and Y. Yan, Chem. Eng. Technol., 2010, 33, 2021–2028 CrossRef CAS.
- M. Milina, S. Mitchell and J. Pérez-Ramírez, Catal. Today, 2014, 235, 176–183 CrossRef CAS.
- Y. Liu, Z. Li, J. J. Leahy and W. Kwapinski, Energy Fuels, 2015, 29, 3691–3698 CrossRef CAS.
- X. Jiang and N. Ellis, Energy Fuels, 2010, 24, 1358–1364 CrossRef CAS.
- X. Junming, J. Jianchun, S. Yunjuan and L. Yanju, Biomass Bioenergy, 2008, 32, 1056–1061 CrossRef.
- N. M. Bennett, S. S. Helle and S. J. B. Duff, Bioresour. Technol., 2009, 100, 6059–6063 CrossRef CAS PubMed.
- R. N. Patel, S. Bandyopadhyay and A. Ganesh, Energy, 2011, 36, 1535–1542 CrossRef CAS.
- M. M. Ishola, A. Jahandideh, B. Haidarian, T. Brandberg and M. J. Taherzadeh, Bioresour. Technol., 2013, 133, 68–73 CrossRef CAS PubMed.
- F. W. Bai, W. A. Anderson and M. Moo-Young, Biotechnol. Adv., 2008, 26, 89–105 CrossRef CAS PubMed.
- B. Dien, M. Cotta and T. Jeffries, Appl. Microbiol. Biotechnol., 2003, 63, 258–266 CrossRef CAS PubMed.
- D. Yuan, K. Rao, P. Relue and S. Varanasi, Bioresour. Technol., 2011, 102, 3246–3253 CrossRef CAS PubMed.
- P. C. Munasinghe and S. K. Khanal, Bioresour. Technol., 2010, 101, 5013–5022 CrossRef CAS PubMed.
- A. Ahmed and R. S. Lewis, Biotechnol. Bioeng., 2007, 97, 1080–1086 CrossRef CAS PubMed.
- D. R. Petrolia, S. Bhattacharjee, D. Hudson and C. W. Herndon, Energ. Econ., 2010, 32, 121–128 CrossRef.
- P. Bajpai, in Advances in Bioethanol, Springer, India, 2013, ch. 4, pp. 55–66, DOI:10.1007/978-81-322-1584-4_4.
- T. C. C. d. Melo, G. B. Machado, C. R. P. Belchior, M. J. Colaço, J. E. M. Barros, E. J. de Oliveira and D. G. de Oliveira, Fuel, 2012, 97, 796–804 CrossRef.
- N. Savage, Nature, 2011, 474, S9–S11 CrossRef CAS PubMed.
- A. K. Agarwal, Prog. Energy Combust. Sci., 2007, 33, 233–271 CrossRef CAS.
- N. Chollacoop, P. Saisirirat, S. Sukkasi, M. Tongroon, T. Fukuda, A. Fukuda and S. Nivitchanyong, Appl. Energy, 2013, 102, 112–123 CrossRef CAS.
- S. Fernando and M. Hanna, Energy Fuels, 2004, 18, 1695–1703 CrossRef CAS.
- A. C. Hansen, Q. Zhang and P. W. L. Lyne, Bioresour. Technol., 2005, 96, 277–285 CrossRef CAS PubMed.
- G. Broustail, P. Seers, F. Halter, G. Moréac and C. Mounaim-Rousselle, Fuel, 2011, 90, 1–6 CrossRef CAS.
- J. Swana, Y. Yang, M. Behnam and R. Thompson, Bioresour. Technol., 2011, 102, 2112–2117 CrossRef CAS PubMed.
- Y. Varol, C. Öner, H. F. Öztop and Ş. Altun, Energy Sources, Part A, 2014, 36, 938–948 CrossRef CAS.
- N. S. Shamsul, S. K. Kamarudin, N. A. Rahman and N. T. Kofli, Renewable Sustainable Energy Rev., 2014, 33, 578–588 CrossRef CAS.
- A. Demirbas, Energy Sources, Part A, 2008, 30, 565–572 CrossRef CAS.
- M. Usman and W. M. A. W. Daud, RSC Adv., 2015, 5, 21945–21972 RSC.
- R. Campo, P. Durán, J. Plou, J. Herguido and J. A. Peña, J. Power Sources, 2013, 242, 520–526 CrossRef CAS.
- M. Balat, M. Balat, E. Kırtay and H. Balat, Energy Convers. Manage., 2009, 50, 3158–3168 CrossRef CAS.
- K. Mazloomi and C. Gomes, Renewable Sustainable Energy Rev., 2012, 16, 3024–3033 CrossRef CAS.
- A. Demirbas, Energy Sources, Part A, 2010, 32, 1342–1354 CrossRef CAS.
- M. Ni, D. Y. C. Leung, M. K. H. Leung and K. Sumathy, Fuel Process. Technol., 2006, 87, 461–472 CrossRef CAS.
- H. Argun and F. Kargi, Int. J. Hydrogen Energy, 2011, 36, 7443–7459 CrossRef CAS.
- M. Balat and M. Balat, Int. J. Hydrogen Energy, 2009, 34, 3589–3603 CrossRef CAS.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef CAS PubMed.
- G. A. Deluga, J. R. Salge, L. D. Schmidt and X. E. Verykios, Science, 2004, 303, 993–997 CrossRef CAS PubMed.
- S. Sarkar and A. Kumar, Bioresour. Technol., 2010, 101, 7350–7361 CrossRef CAS PubMed.
- W. Zhang, Fuel Process. Technol., 2010, 91, 866–876 CrossRef CAS.
- Z. Azizi, M. Rezaeimanesh, T. Tohidian and M. R. Rahimpour, Chem. Eng. Process., 2014, 82, 150–172 CrossRef CAS.
- A. M. Rouhi, Chem. Eng. News, 1995, 73, 37–39 Search PubMed.
- C. Arcoumanis, C. Bae, R. Crookes and E. Kinoshita, Fuel, 2008, 87, 1014–1030 CrossRef CAS.
- J. Chang, Y. Fu and Z. Luo, Biomass Bioenergy, 2012, 39, 67–72 CrossRef CAS.
- A. T. Aguayo, J. Ereña, D. Mier, J. M. Arandes, M. Olazar and J. Bilbao, Ind. Eng. Chem. Res., 2007, 46, 5522–5530 CrossRef CAS.
- D. M. Brown, B. L. Bhatt, T. H. Hsiung, J. J. Lewnard and F. J. Waller, Catal. Today, 1991, 8, 279–304 CrossRef CAS.
- K. T. Lee, S. Lim, Y. L. Pang, H. C. Ong and W. T. Chong, Prog. Energy Combust. Sci., 2014, 45, 54–78 CrossRef.
- M. Hasheminejad, M. Tabatabaei, Y. Mansourpanah, M. K. far and A. Javani, Bioresour. Technol., 2011, 102, 461–468 CrossRef CAS PubMed.
- A. Demirbaş, Energy Convers. Manage., 2002, 43, 2349–2356 CrossRef.
- P. D. Patil and S. Deng, Fuel, 2009, 88, 1302–1306 CrossRef CAS.
- U. Addepally and C. Thulluri, Fuel, 2015, 159, 935–942 CrossRef CAS.
- IEA, Sustainable production of second-generation biofuels: potential and perspectives in major economies and developing countries, International Energy Agency (IEA/OECD), Paris, 2010 Search PubMed.
- R. E. H. Sims, W. Mabee, J. N. Saddler and M. Taylor, Bioresour. Technol., 2010, 101, 1570–1580 CrossRef CAS PubMed.
- J. Saddler, W. Mabee, R. Simms and M. Taylor, in Forests in Development: A Vital Balance, ed. T. Schlichter and L. Montes, Springer, Netherlands, 2012, ch. 4, pp. 39–51, DOI:10.1007/978-94-007-2576-8_4.
- S. Gold and S. Seuring, J. Cleaner Prod., 2011, 19, 32–42 CrossRef.
- D. Andress, T. D. Nguyen and S. Das, Energy Sustainable Dev., 2011, 15, 117–136 CrossRef CAS.
- F. B. Zhan, X. Chen, C. E. Noon and G. Wu, Biomass Bioenergy, 2005, 28, 295–306 CrossRef.
- J. R. Hess, C. T. Wright and K. L. Kenney, Biofuels, Bioprod. Biorefin., 2007, 1, 181–190 CrossRef CAS.
- M. Langholtz, R. Graham, L. Eaton, R. Perlack, C. Hellwinkel and D. G. De La Torre Ugarte, Energy Policy, 2012, 41, 484–493 CrossRef.
- R. F. Jeffers, J. J. Jacobson and E. M. Searcy, Energy Policy, 2013, 52, 249–263 CrossRef.
- M. M. Rahman and J. V. Paatero, Biomass Bioenergy, 2012, 47, 153–163 CrossRef.
- J. Sun, J. Chen, Y. Xi and J. Hou, J. Cleaner Prod., 2011, 19, 121–128 CrossRef.
- J. Singh, B. S. Panesar and S. K. Sharma, Biomass Bioenergy, 2010, 34, 483–488 CrossRef.
- V. Balan, D. Chiaramonti and S. Kumar, Biofuels, Bioprod. Biorefin., 2013, 7, 732–759 CrossRef CAS.
- Z. Miao, Y. Shastri, T. E. Grift, A. C. Hansen and K. C. Ting, Biofuels, Bioprod. Biorefin., 2012, 6, 351–362 CrossRef CAS.
- H. L. Wakeley, C. T. Hendrickson, W. M. Griffin and H. S. Matthews, Environ. Sci. Technol., 2009, 43, 2228–2233 CrossRef CAS PubMed.
- D. Yue, F. You and S. W. Snyder, Comput. Chem. Eng., 2014, 66, 36–56 CrossRef CAS.
- M. Santamaría and D. Azqueta, Renewable Sustainable Energy Rev., 2015, 50, 1415–1424 CrossRef.
- C. B. Granda, L. Zhu and M. T. Holtzapple, Environ. Prog., 2007, 26, 233–250 CrossRef CAS.
- H. Chen and X. Fu, Renewable Sustainable Energy Rev., 2016, 57, 468–478 CrossRef CAS.
- T. R. Brown and R. C. Brown, Biofuels, Bioprod. Biorefin., 2013, 7, 235–245 CrossRef CAS.
- RFA, Going Global-2015 ethanol Industry Outlook, Renewable Fuels Association (RFA), Washington DC, 2015 Search PubMed.
- AFDC, Global Ethanol Production, http://www.afdc.energy.gov/data/10331, accessed January 30, 2016, 2016.
- UNEP, Renewables 2015 Global Status Report, Paris, France, 2015 Search PubMed.
- A. d. R. Pinho, M. B. B. de Almeida, F. L. Mendes, V. L. Ximenes and L. C. Casavechia, Fuel Process. Technol., 2015, 131, 159–166 CrossRef CAS.
- I. Graça, J. M. Lopes, H. S. Cerqueira and M. F. Ribeiro, Ind. Eng. Chem. Res., 2013, 52, 275–287 CrossRef.
- J. G. Speight, in The Refinery of the Future, ed. J. G. Speight, William Andrew Publishing, Boston, 2011, pp. 315–340, DOI:10.1016/b978-0-8155-2041-2.10010-4.
- J. D. Adjaye and N. N. Bakhshi, Fuel Process. Technol., 1995, 45, 161–183 CrossRef CAS.
- E. Gnansounou and A. Dauriat, Bioresour. Technol., 2010, 101, 4980–4991 CrossRef CAS PubMed.
- P. Tunå and C. Hulteberg, Fuel, 2014, 117, 1020–1026 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |