
Hydrolytic stability of end-linked hydrogels from PLGA–PEG–PLGA macromonomers terminated by α,ω-itaconyl groups†
L. Michlovská*a,
L. Vojtová*ab,
O. Humpac,
J. Kučeríkd,
J. Žídeka and
J. Jančářab
aCEITEC – Central European Institute of Technology, Brno University of Technology, Technická 3058/10, 616 00, Brno, Czech Republic. E-mail: lenka.michlovska@ceitec.vutbr.cz; lucy.vojtova@ceitec.vutbr.cz
bSCITEG, a.s., Brno, Czech Republic
cCEITEC – Central European Institute of Technology, Masaryk University, Kamenice 753/5, 625 00 Brno, Czech Republic
dUniversity of Koblenz-Landau, Institute of Environmental Sciences, Soil and Environmental Chemistry, Forstr. 7, 76829 Landau, Germany
First published on 4th February 2016
Abstract
Biodegradable amphiphilic PLGA–PEG–PLGA triblock copolymers end-terminated with itaconic acid (ITA) having reactive double bonds were synthesized by ring opening polymerization. The prepared α,ω-itaconyl-PLGA–PEG–PLGA telechelic macromonomers were additionally covalently crosslinked under an inert atmosphere by blue light irradiation without the use of a further cross-linker resulting in end-linked polymeric networks. The effects of the ITA amount attached to the α,ω-itaconyl-PLGA–PEG–PLGA copolymers and the crosslinking time on swelling behaviours and hydrolytic stability of the prepared well-defined polymeric network were investigated. Physicochemical properties were characterized by proton and carbon nuclear magnetic resonance spectroscopy (1H NMR, 13C NMR), proton nuclear magnetic resonance relaxometry, attenuated total reflectance Fourier transformed infrared spectroscopy (ATR-FTIR) and thermogravimetric analysis (TGA). It was found that the hydrolytic stability of ITA modified PLGA–PEG–PLGA end-linked hydrogels enhances with both increasing the time of crosslinking and the amount of double bonds attached to α,ω-itaconyl-PLGA–PEG–PLGA polymer chains. In comparison with the original un-crosslinked α,ω-itaconyl-PLGA–PEG–PLGA copolymer, the hydrolytic stability of the end-linked hydrogels significantly increased. Three kinds of water fractions (unbound, weakly and strongly bonded) were determined by proton NMR relaxometry in hydrogels containing 63 mol% of ITA crosslinked for 40 minutes. Even for hydrogels surviving 32 days in water the NMR relaxometry showed structural collapse of the hydrogel probably due to breaking of end-linked nodes followed by hydrolysis faster than water diffusion after day 15 of immersion. End-linked α,ω-itaconyl-PLGA–PEG–PLGA hydrogels can be used in medical, biological or tissue engineering applications.
Introduction
During the last two decades, biodegradable polymers and copolymers have met with increasing interest in medicine and biology, particularly in the fields of tissue engineering, cell therapies, wound treatment as well as drug carriers. The most frequently used are linear aliphatic polyesters such as polyglycolide (PGA), polylactide (PLA) and their statistical copolymer of polylactide-co-polyglycolide (PLGA), whose ester bonds undergo homogenous hydrolysis. The degradation rate of PLGA copolymers depends on their chemical structure, the homopolymer ratio of PLA and PGA and their molecular weight. Hydrophobic PLGA copolymer is often chemically modified by hydrophilic poly(ethylene glycol) (PEG), which is not detectable by immune system and helps to dissolve PLGA in aqueous solutions. Specifically, amphiphilic triblock PLGA–PEG–PLGA copolymer is biocompatible, biodegradable, resorbable, thermosensitive and after hydrolysis at physiological conditions to lactic and glycolic acids undergo degradation via the Krebs cycle to non-toxic products (up to a water and carbon dioxide).1,2Aqueous solution of PLGA–PEG–PLGA copolymer is in a sol (liquid) phase at room temperature and turned to a gel at human body temperature.3,4 This copolymer is commercially known as an injectable drug carrier ReGel® releasing insulin for treatment diabetes mellitus Type I5 or in combination with paclitaxel is known as OncoGel® used for targeted cancer therapy.6 However, its use in medicine is limited due to the fact that thermogelling sol–gel process caused by hydrophobic interactions is reversible, and copolymers have a low degree of functionality (they contain only hydroxyl functional groups). For this reason, much attention is devoted to the modification of the functional groups and chemical (irreversible) cross-linking of these materials. Chemical crosslinking decreases the rate of polymer degradation. The resulting hydrogels are more stable, and therefore degrade more slowly.5,7 These properties are particularly preferred for temporary orthopaedic implants, which gradually degrade during the healing and growth of human bones.8,9
In order to obtain functionalized polymers suitable for chemical end-linking (so called macromonomers), hydroxyl terminated poly(ε-caprolactone) (PCL), polylactide (PLA) and polyglycolide (PGA) have already been modified by maleic anhydride, fumaric acid, acrylate, methacrylic anhydride and triethoxy(3-isocyanatopropyl)silane.10–15
However, the use of itaconic anhydride (ITAn) for polymer end-functionalization has been reported only in the case of PCL.11,16 Chemically end-linked polyesters were formed by curing these PCL macromonomers using thermoinitiators, redox systems or photoinitiators.17–20 Modification of PLGA–PEG–PLGA triblock copolymer by ITA was firstly reported by our group.21
Itaconic anhydride is unsaturated cyclic anhydride and it can be obtained from renewable resources both by distillation of citric acid and pyrolysis of itaconic acid. ITA is produced mainly by fermentation of polysaccharides (e.g. molasses, glucose) using bacteria of Aspergillus terreus.22 It is generally known that itaconic anhydride passes to nontoxic degradation products under physiological conditions, when it initially hydrolyses to ITA in water. PLGA–PEG–PLGA copolymer was modified by itaconic anhydride in “one-pot” reaction. The itaconic anhydride is bonded through the ring-opening reaction and formed itaconic acid at the ends of copolymer brings both reactive double bonds suitable for chemical crosslinking and functional carboxylic acid groups essential for the reaction with biologically active material.
This paper is focused on the chemical crosslinking of α,ω-itaconyl-PLGA–PEG–PLGA (ITA/PLGA–PEG–PLGA/ITA) macromonomers by blue light without added crosslinker. The aim is to form end-linked well-organized gel network from water-soluble PLGA–PEG–PLGA copolymer, which hydrolytic stability can be controlled by the amount of bonded functional double bond of itaconic acid and the crosslinking time. Effects of the ITA amount bounded to the ends of PLGA–PEG–PLGA copolymer and macromonomer crosslinking time on swelling properties and hydrolytic stability of prepared end-linked hydrogels were investigated.
Experimental procedure
Materials
D,L-Lactide (LA, ≥99.9%) and glycolide (GA, ≥99.9%) were supplied by Polysciences (Pennsylvania) and itaconic anhydride (ITAn 97%) by FLUKA (Switzerland). Poly(ethylene glycol) (PEG, Mn = 1500 g mol−1), Sn(II)2-ethylhexanoate (95%), camphorquinone (1,7,7-trimethyl-bicyclo(2,2,1)-heptan-2,3-dione, CQ, 97.0%) and 2-(dimethylamino) ethyl methacrylate (98.0%) were purchased from Sigma-Aldrich (Germany). Phosphate buffered saline with pH equal to 7.48 (PBS 7.48) was prepared using NaCl, KCl, Na2HPO4, KH2PO4 by adjusting pH either with NaOH (c = 1 mol dm−3) or HCl (c = 1 mol dm−3). Ultrapure water (Type 1 according to ISO 3696) was prepared using Millipore purification system (MilliQ Academic, Millipore, France).Synthesis of α,ω-itaconyl-PLGA–PEG–PLGA macromonomer
The α,ω-itaconyl-PLGA–PEG–PLGA triblock copolymer (Scheme 1) with weight ratio of PLGA/PEG = 2.5 and molar ratio of PLA/PGA = 3.0 was synthesized in “one pot” via ring opening polymerization (ROP) method in a bulk under nitrogen atmosphere as described in detail elsewhere.19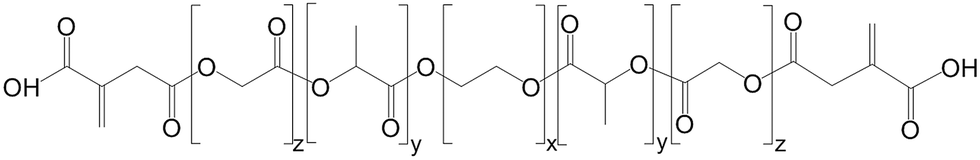 | ||
| Scheme 1 α,ω-Itaconyl-PLGA–PEG–PLGA triblock copolymer. | ||
Shortly, PEG, LA and GA were homogenized at 130 °C followed by injecting Sn(II)2-ethylhexanoate. Reaction ran over 3 hours and then PLGA–PEG–PLGA copolymer was modified in second step by ring-opening reaction of itaconic anhydride (2.5 molar ratio) at 110 °C for 1.5 hour in a bulk. Unreacted itaconic anhydride was removed from copolymer by purification and decantation at 80 °C in water. Modification by itaconic anhydride brings both functional carboxylic groups and double bonds to the ends of copolymer thus forming functional macromonomer.
Preparation of end-linked hydrogels
Chemically end-linked gels were prepared in the presence of camphorquinone initiator with maximum absorption wavelength at 468 nm and 2-(dimethylamino)ethyl methacrylate (DMAEM) used as catalyst without the use of solvent. Samples were chemically crosslinked by LED polymerization lamp with wavelength between 430–490 nm and blue light intensity region of 800–1200 mW cm−2. 0.3 g of α,ω-itaconyl-PLGA–PEG–PLGA macromonomer was placed on thermoblock set up to 60 °C. 5.6 mg of camphorquinone and 4.5 μL of 2-(dimethylamino)ethyl methacrylate were added to the sample. Sample was dosed into the mould and chemically crosslinked by blue light. Crosslinking time was either 5 or 40 minutes for copolymer with 37 mol% of ITA and 40 minutes for copolymer with 63 mol% of ITA as described in Table 1. Samples were dried in vacuum oven at laboratory temperature for 8 hours.| Sample description | ITA [mol%] | Crosslinking time [min] |
|---|---|---|
| 37ITAx0 | 37 | 0 |
| 63ITAx0 | 63 | 0 |
| 37ITAx5 | 37 | 5 |
| 37ITAx40 | 37 | 40 |
| 63ITAx40 | 63 | 40 |
Characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), scans (128), recycle delay (12 s) and echo time (1 ms) were kept constant. The obtained induction decay curves were fitted using eqn (1).
000), scans (128), recycle delay (12 s) and echo time (1 ms) were kept constant. The obtained induction decay curves were fitted using eqn (1).
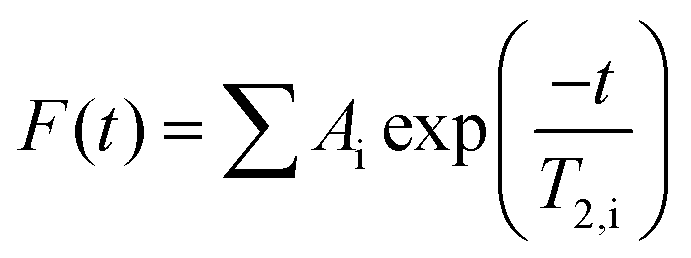 | (1) |
Swelling and hydrolytic stability
Samples were immersed in an excess of ultrapure water with pH of 6.5 at room temperature. Swelling kinetics was followed gravimetrically by recording weight increments as function of time until equilibrium was reached. After removing the swollen samples from the solutions at regular intervals, they were dried superficially with filter paper, weighed and returned to the solution. The ultrapure water was changed every 24 hours. Water content (WC) was calculated using eqn (2)
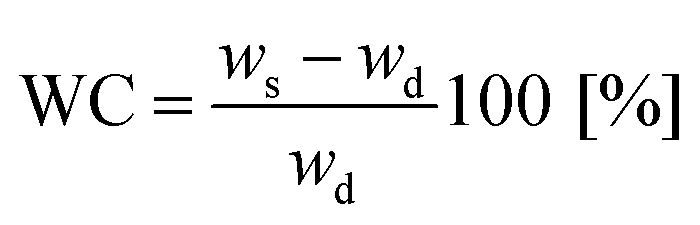 | (2) |
Results and discussion
Functionalization efficacy of PLGA–PEG–PLGA copolymers by ITA
PLGA–PEG–PLGA triblock copolymers with PLGA/PEG weight ratio equal to 2.5 and PLA/PGA molar ratio equal to 3.0 were synthesized via ring opening polymerization and subsequently functionalized with itaconic anhydride (ITAn) in order to prepare α,ω-itaconyl-PLGA–PEG–PLGA macromonomers.Molecular weights Mn and molar composition of samples were determined by 1H NMR spectroscopy (Fig. 1) from integrals of characteristic proton intensities of lactic acid (O(CH3)C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) O) in a range between δ = 5.1–5.3 ppm (m, signal 3) and (OC(C3)CHO) protons at δ = 1.4–1.65 ppm (overlapped dublets, signal 4), glycolic acid (OC2O) at δ = 4.6–4.9 ppm (area of overlapped singlets, signal 5), PEG (OC2C2O) at δ = 3.55–3.75 ppm (several overlapped peaks, signal 1). Signal at δ = 4.20–4.35 ppm (overlapped peaks, signal 2) corresponded to proton from –CH2– group at bond between PEG and PLA (OC2C2O).
O) in a range between δ = 5.1–5.3 ppm (m, signal 3) and (OC(C3)CHO) protons at δ = 1.4–1.65 ppm (overlapped dublets, signal 4), glycolic acid (OC2O) at δ = 4.6–4.9 ppm (area of overlapped singlets, signal 5), PEG (OC2C2O) at δ = 3.55–3.75 ppm (several overlapped peaks, signal 1). Signal at δ = 4.20–4.35 ppm (overlapped peaks, signal 2) corresponded to proton from –CH2– group at bond between PEG and PLA (OC2C2O).
 | ||
| Fig. 1 1H NMR spectrum of original and α,ω-itaconyl-PLGA–PEG–PLGA copolymers (a) 37ITAx0, (b) 63ITAx0, where 1 corresponds to proton in PEG, 2 – proton from –CH2– group at bond between PEG and PLA, 3 and 4 – protons of lactic acid, 5 – CH2 group at bond between copolymer and attached itaconic acid, 6 – protons in CH2 group at bond between copolymer and attached itaconic acid, 7 – protons from itaconic acid on the double bond. | ||
The calculated amount of ITA in prepared samples was 37 and 63 mol% (sample 37ITAx0 and 63ITAx0). Degree of functionalization equal to 0.74 or 1.26, respectively, depended on the ITA purification step prior the modification. Sublimated ITA was evacuated at room temperature prior the reaction with PLGA–PEG–PLGA for either 1 or 2 hours, respectively, to obtain different functionalization efficacy.
Amount of end-capped ITA was determined from integrals of characteristic proton signals of itaconic acid double bonds (OC(CH2)CC2COOH) at δ = 5.8–5.9 ppm (bs, signal 7b) and δ = 6.35–6.5 ppm (bs, signal 7a). Bonding of ITA to the end of copolymers was proved by peak at δ = 3.40–3.44 ppm (overlapped singlets, signal 6)24,25 corresponding to proton from –CH2– group at bond between copolymer and attached itaconic acid.
Molecular weights, chemical composition (LA/GA molar ratio and PLGA/PEG weight ratio) measured by 1H NMR spectroscopy are listed in Table 2. Molecular weights and composition ratios were in very good agreement with theoretical values.
| Sample | PLGA/PEG [wt/wt] | LA/GA [mol/mol] | Mn(NMR) g mol−1 | Mn(theor)/Mn(NMR) |
|---|---|---|---|---|
| 37ITAx0 | 2.47 | 2.77 | 5269 | 0.994 |
| 63ITAx0 | 2.59 | 2.97 | 5384 | 0.975 |
Copolymers were further characterized by 13C NMR spectroscopy (frequency 176.1 MHz) in CDCl3. Representative spectrum is shown at Fig. 2.
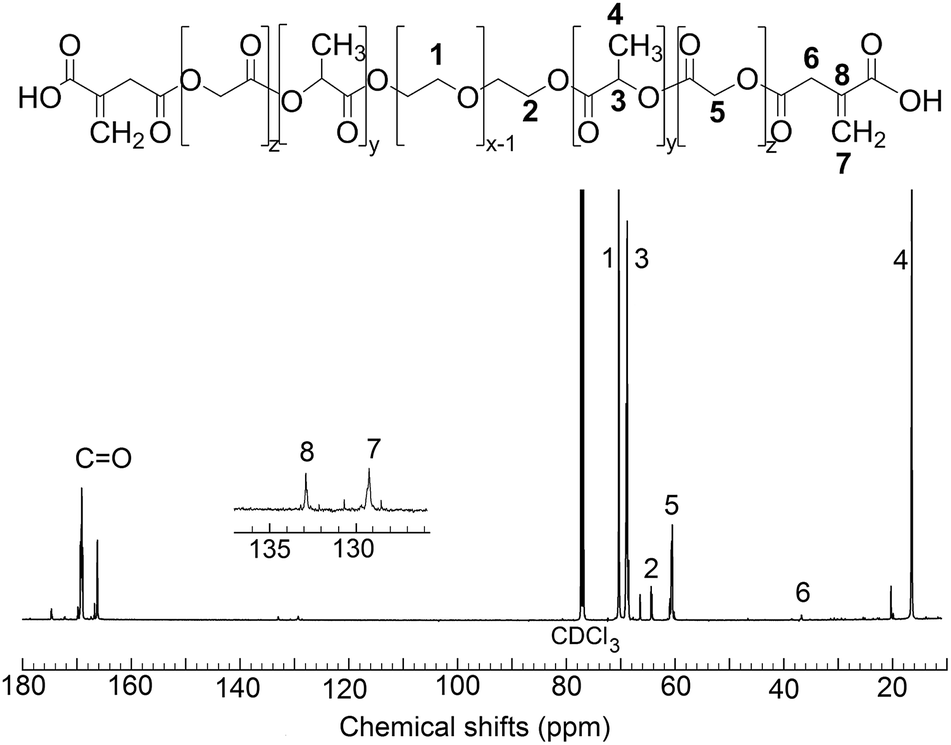 | ||
| Fig. 2 13C NMR spectrum of 63ITAx0, where 1 corresponds to carbon in PEG, 2 – CH2 group at bond between PEG and PLA, 3 and 4 – carbons in lactic acid, 5 is carbon in glycolic acid, 6 – CH2 group at bond between copolymer and attached itaconic acid, 7a and 7b – carbons in itaconic acid double bonds. | ||
It was confirmed that signal 4 at δ = 16.4 ppm (q) corresponded to (O–(![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3)CHO) carbon in lactic acid, signal 6 at δ = 26.76 ppm (t) to CH2– group at bond between copolymer and attached itaconic acid, signal 5 at δ = 60.4 ppm (t) to (OH2O) carbon in glycolic acid, signal 2 at δ = 64.3 ppm (t) to (OCOH2CH2) carbon from –CH2– group at bond between PEG and PLA, peak 3 at δ = 68.9 ppm (d) to (OC(CH3)HO) carbon in lactic acid, peak 1 at δ = 70.3 ppm (t) to (OCH2H2O) in PEG and peak at δ = 169.7 ppm to carbonyls. ITA was determined from peak 8 at δ = 132.9 ppm (s) corresponded to (OC(CH2)CH2COOH) carbon and peak 7 to
H3)CHO) carbon in lactic acid, signal 6 at δ = 26.76 ppm (t) to CH2– group at bond between copolymer and attached itaconic acid, signal 5 at δ = 60.4 ppm (t) to (OH2O) carbon in glycolic acid, signal 2 at δ = 64.3 ppm (t) to (OCOH2CH2) carbon from –CH2– group at bond between PEG and PLA, peak 3 at δ = 68.9 ppm (d) to (OC(CH3)HO) carbon in lactic acid, peak 1 at δ = 70.3 ppm (t) to (OCH2H2O) in PEG and peak at δ = 169.7 ppm to carbonyls. ITA was determined from peak 8 at δ = 132.9 ppm (s) corresponded to (OC(CH2)CH2COOH) carbon and peak 7 to ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 at δ = 129.2 ppm (t) to (OC(CH2)CH2COOH) carbon.
CH2 at δ = 129.2 ppm (t) to (OC(CH2)CH2COOH) carbon.
Presence of itaconic acid in PLGA–PEG–PLGA copolymer was confirmed by two dimensional 1H-13C HMBC (heteronuclear multiple bond correlation) experiment (Fig. 3). Protons of itaconic acid from peaks 7 and 8 provide correlation through three linkages with proton at bond between copolymer and attached itaconic acid (peak 6).
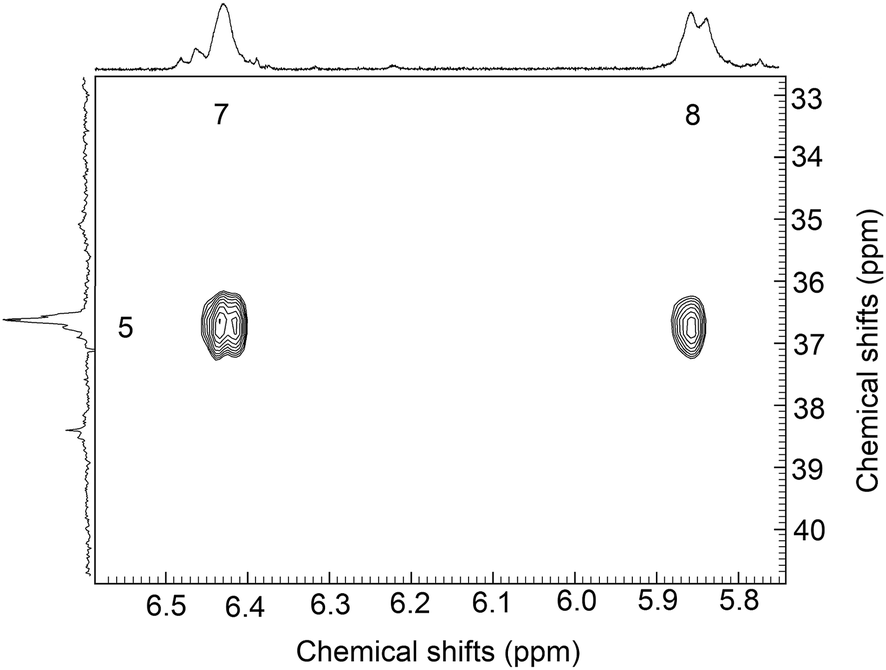 | ||
| Fig. 3 Detail in two dimensional 1H-13C HMBC spectrum of sample 63ITAx0 and attached itaconic acid. Cross peaks correspond long-range heteronuclear correlation between carbon 6 and protons on the carbon 7 attached itaconic acid. | ||
End-linking of α,ω-itaconyl-PLGA–PEG–PLGA macromonomers
Chemical end-linking of α,ω-itaconyl-PLGA–PEG–PLGA macromonomers was proceeded by LED blue light with wavelength between 430–490 nm. End-linking efficacy has been evaluated with a view of the effect of crosslinking time and the amount of bonded ITA in α,ω-itaconyl-PLGA–PEG–PLGA macromonomers. Crosslinking proceeded in the presence of D,L-camphorquinone catalyst (1,7,7-trimethy-bicyclo(2,2,1)-heptane-2,3-dione) and tertiary amine 2-(dimethylamino) ethyl methacrylate without the use of crosslinking agent. The mechanism of VIS-initiated crosslinking is a radical reaction (Scheme 2).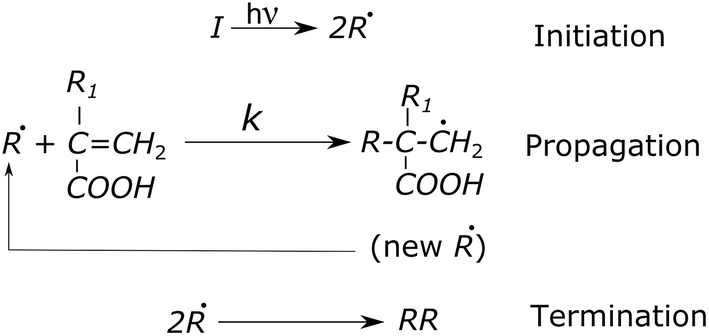 | ||
| Scheme 2 Mechanism of covalent crosslinking; I is initiator, R is a radical, k is a rate constant of propagation. | ||
The quantum chemical simulation gives the indication that the radical attacks the double CC bond in the itaconic acid next to the COOH group. The highest occupied molecular orbital (HOMO) was calculated using Density Functional Theory (DFT) Kohn–Sham method with Becke's exchange functional and 3-parameter, Lee–Yang–Parr correlation function (KS, B3LYP). The basis set used was 6-31G.
The visualization of HOMO orbital in vicinity of double bond (DB) is presented in Fig. 4. The radical – a single occupied molecular orbital (SOMO) – is illustrated as a green ball. A sigma bond created by overlapping SOMO radical and HOMO double bond (Fig. 4 upper-right) leads to arising new SOMO orbital (available for chain reaction).
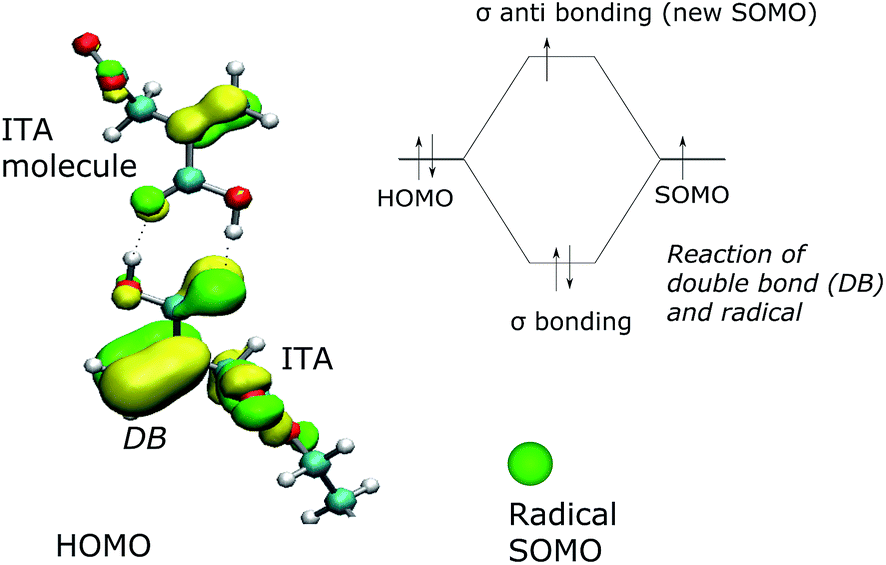 | ||
| Fig. 4 The quick phase of creation of sigma bond after collision of radical and double bond (DB) in propagation step. Rate of this reaction is driven by frequency of such collisions; HOMO – the highest occupied molecular orbital, SOMO – single occupied molecular orbital. | ||
The influence of the amount of bonded ITA and crosslinking time on the structural changes determined by attenuated total reflectance Fourier transformed infrared spectroscopy (ATR-FTIR) is shown in Fig. 5.
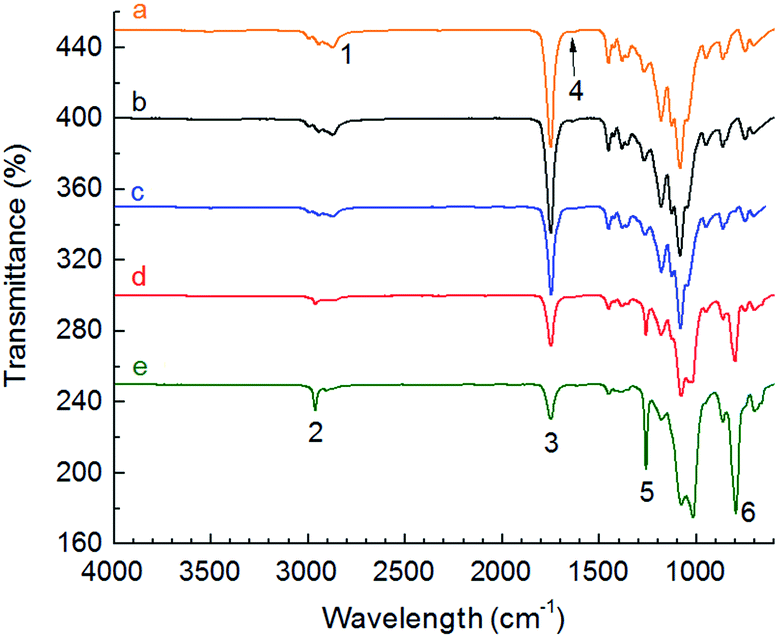 | ||
| Fig. 5 Infrared spectra of original and α,ω-itaconyl-PLGA–PEG–PLGA copolymers, where (a) 37ITAx0, (b) 63ITAx0, (c) 37ITAx5, (d) 37ITAx40 and (e) 63ITAx40. | ||
The samples exhibited changes in intensities at wavelength of 2900 cm−1 (peak 1) corresponded to –OH in carboxylic group. Original uncrosslinked sample contains conjugated double bonds with carboxylic acid groups. Peak of –OH is broad and it interferes with –CH bond vibration. Chemically crosslinked copolymers contain separated peak at 2965 cm−1 (peak 2), which has single C–C bond neat the –COOH group. That can indicate, the increasing time of crosslinking and amount of ITA leads to transformation of double CC bond to the single C–C bond. Magnification of peaks 1 and 2 in a range of 2800 to 3100 cm−1 is shown at Fig. S1.† Peak 3 at 1750 cm−1 corresponds to CO group and peak 5 to C–O stretch. Decrease of double bonds in itaconic acid (RRCCH2) at 1640 cm−1 (peak 4) and formation of new bonds (RRC–CHR) at 795 cm−1 (peak 6) were observed after crosslinking (Fig. 6).
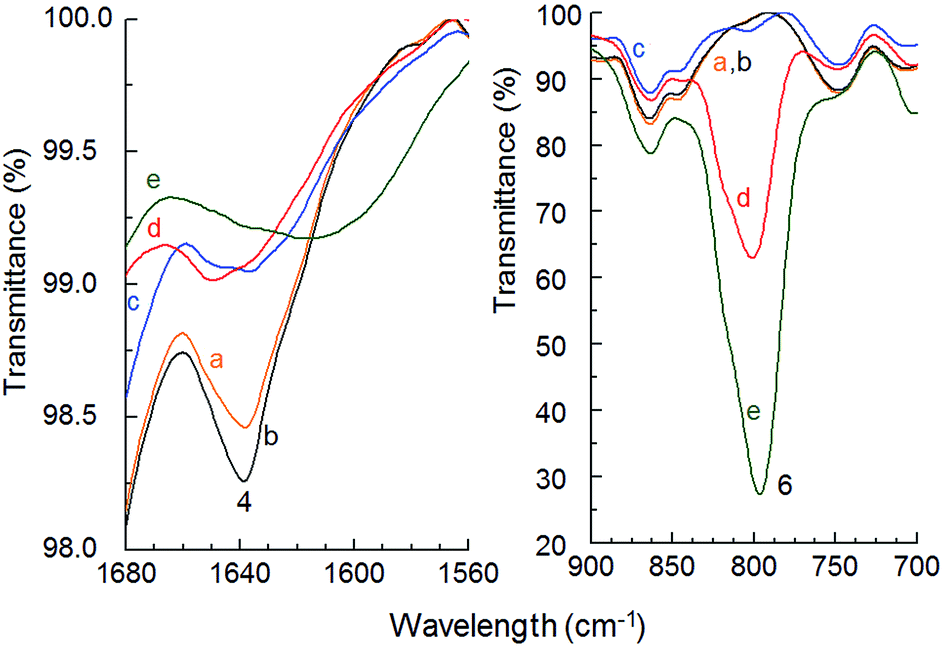 | ||
| Fig. 6 Magnification of peaks in region 1560–1680 and 700–900 cm−1, where (a) 37ITAx0, (b) 63ITAx0, (c) 37ITAx5, (d) 37ITAx40 and (e) 63ITAx40. | ||
Content of double bonds in the hydrogels and crosslinking degree of double bonds were determined by 1H NMR spectroscopy. Table 3 confirms high crosslinking degree (more than 93%), even after first 5 min of crosslinking, as it was seen from the ATR-FTIR measurement of double bonds above. However, the sample 63ITAx40 showed almost 98% of double bond conversion.
| Sample description | Double bonds before/after crosslinking [mol%] | Crosslinking degree [%] |
|---|---|---|
| 37ITAx0 | 37.0 | — |
| 63ITAx0 | 63.0 | — |
| 37ITAx5 | 2.3 | 93.8 |
| 37ITAx40 | 2.1 | 94.3 |
| 63ITAx40 | 1.3 | 97.9 |
Thermal stability of original and crosslinked copolymers were studied by thermogravimetric analysis. As can be seen, crosslinked samples with chemical junctions had also better thermal stability (Fig. 7 and S2†).
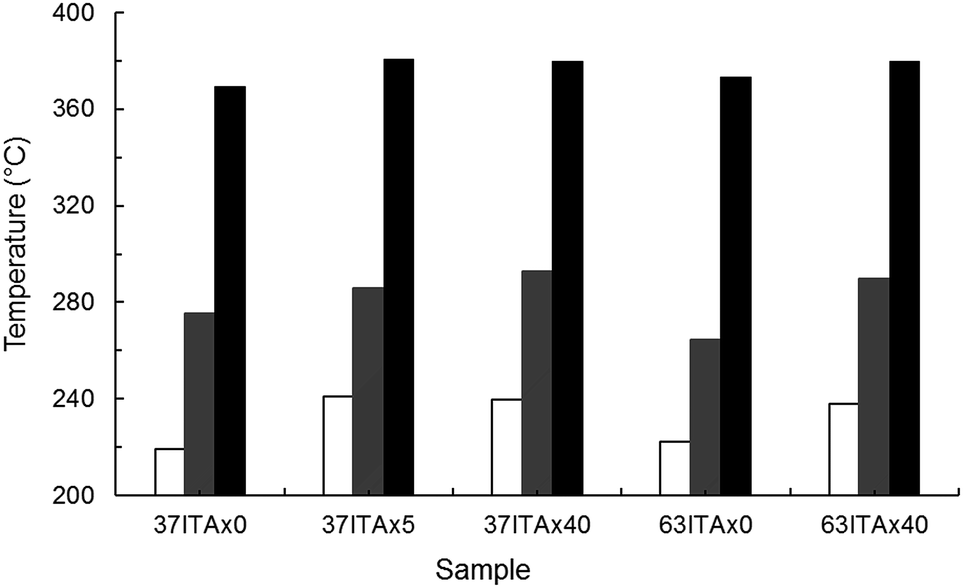 | ||
| Fig. 7 Thermal stability of original and crosslinked copolymers by thermogravimetric analysis, where white area corresponded to Tonset, grey area to Tdm1 and black area to Tdm2. | ||
Crosslinked samples started to degrade approx. from 239 °C, in comparison with 37ITAx0 and 63ITAx0, which degraded approx. at 220 °C (Tonset). For the copolymer 63ITAx40, noticeably increased the temperature of the maximum rate of degradation of esters chains (Tdm1) and slightly decomposition of ethers chains (Tdm2) from original 264 to 290 °C and 373 to 380 °C, respectively. Temperature at maximum rate Tdm1 increased from 276 to 286 and 293 °C for samples 37ITAx5, 37ITAx40 and decomposition of ether bonds (Tdm2) from 369 to 381 and 380 °C, respectively.
Hydrolytic stability of end-linked hydrogels
Hydrolytic stability and degradation rate of samples were measured in ultrapure water at laboratory temperature. From the graph shown in Fig. 8 it is apparent that sample 37ITAx0 absorbed water very quickly at the beginning of the swelling (up to 2547%) followed by complete dissolving in 48 hours. In contrast, the same sample crosslinked for 5 minutes (37ITAx5) by LED blue light exhibited gradual diffusion of water into the polymer network during the first 7 days with slow increasing water uptake up to 574%. Water firstly hydrated hydrophilic groups of PEG and carboxylic end groups. Increasing time causes hydrolysis of part of ester bonds accompanied by reducing the network density causing pores in the structure accompanied by the absorption of extra large amount of water up to 1103%. Subsequently, network nodes were broken and hydrolysis of rest of PLGA ester bonds began together with the release of lactic and glycolic acid. For this reason dissolution of sample was faster than diffusion and sample was completely dissolved in 10 days from the beginning of the swelling.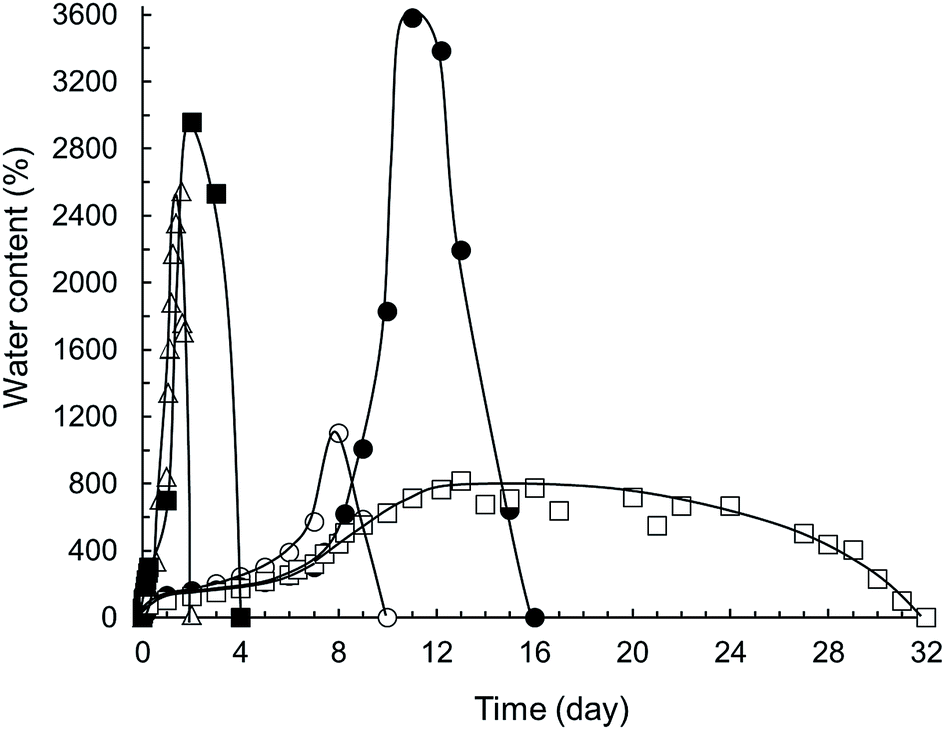 | ||
| Fig. 8 Swelling and degradation of α,ω-itaconyl-PLGA–PEG–PLGA samples in ultrapure water at 25 °C, where △ is sample 37ITAx0, ○ 37ITAx5, ● 37ITAx40, ■ 63ITAx0 and □ 63ITAx40. | ||
If the sample was crosslinked longer time (40 minutes), the crosslinking density increased with improving of a water stability and keeping the polymer chain together up to a maximum of 3581% of water content (weight increased almost 37×). Sample started to dissolve after 11 days and it whole degraded at 16th day. Different situation happened when the α,ω-itaconyl-PLGA–PEG–PLGA macromonomer end-capped with 63 mol% of ITA was crosslinked by blue lamp for 40 minutes. This sample followed the swelling tendency by the same route as sample 37ITAx40 up to 8 day. After this time, there has been only slight increase of water amount (813%). In the period between the 13th and 20th day, there was observed a balance between diffusion and hydrolysis of the hydrogel followed by slow dissolution and partial degradation, since the sample involved a lot of crosslinking groups supporting high crosslinking density. Sample 63% ITA cross. 40 min disintegrated in 32nd days. From the Fig. 8 it is apparent that hydrolytic stability of ITA/PLGA–PEG–PLGA/ITA hydrogels (degradation) is possible to affect both by the time of crosslinking and the amount of bonded ITA.
Non-crosslinked, end-crosslinked hydrogels and samples after swelling and degradation were characterized by gel permeation chromatography (Table 4). Molecular weights and PDI greatly reduced with increasing amount of itaconic acid, longer crosslinking time and time of hydrolytic stability measurement. The largest difference was observed at the sample 63ITAx40 (Fig. 9). Long hydrolytic stability in water (up to 32 days) (Fig. 8) affected final molecular weight and polydispersity indexes after swelling and degradation measurement. Mn decreased 3.2 times compared to initial value of end-linked hydrogel and PDI increased from 1.56 to 5.10. Similar molecular weights and PDI of original samples before and after swelling show, that both samples were dissolved without degradation.
 | ||
| Fig. 9 GPC chromatograms of hydrogels, 1 – 63ITAx0, 2 – 63ITAx40 and 3 – 63ITAx40 after swelling and degradation. | ||
α,ω-Itaconyl-PLGA–PEG–PLGA copolymer labelled as 37ITAx40 (a) and 63ITAx40 (b) in swelled state at 11th day of swelling are shown at Fig. 10. Copolymer 37ITAx40 was able to absorb more water due to lower network density and the amount of network nodes due to the lower amount of double bonds coming from ITA.
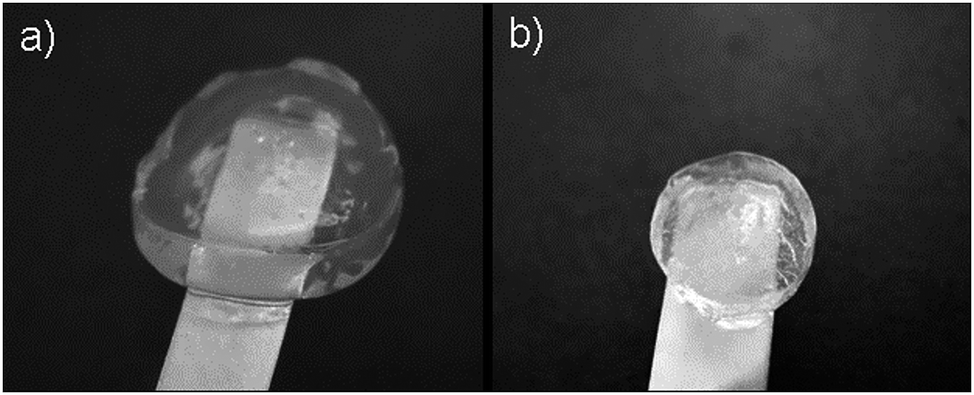 | ||
| Fig. 10 Crosslinked samples (a) 37ITAx40 and (b) 63ITAx40 in swelled state at 11th day. | ||
Swelling and subsequent degradation in ultrapure water gives the quantitative information about total amount of absorbed water. The detail information about the dynamics of relevant processes and abundance of water fractions was obtained from the fitting of free induction decay curves from proton NMR relaxometry measurement using eqn (1). The best fit gave the three-exponential function, which means that system contained (at least) three spin–spin (transversal) relaxation times (T2) corresponding to three water fractions. The relative abundances of those fractions are reflected by the respective amplitudes of fitting (Ai). The results of the successive hydration of 63 mol% ITA are shown at Fig. 11 (fraction abundance) and Fig. 12 (relaxation times).
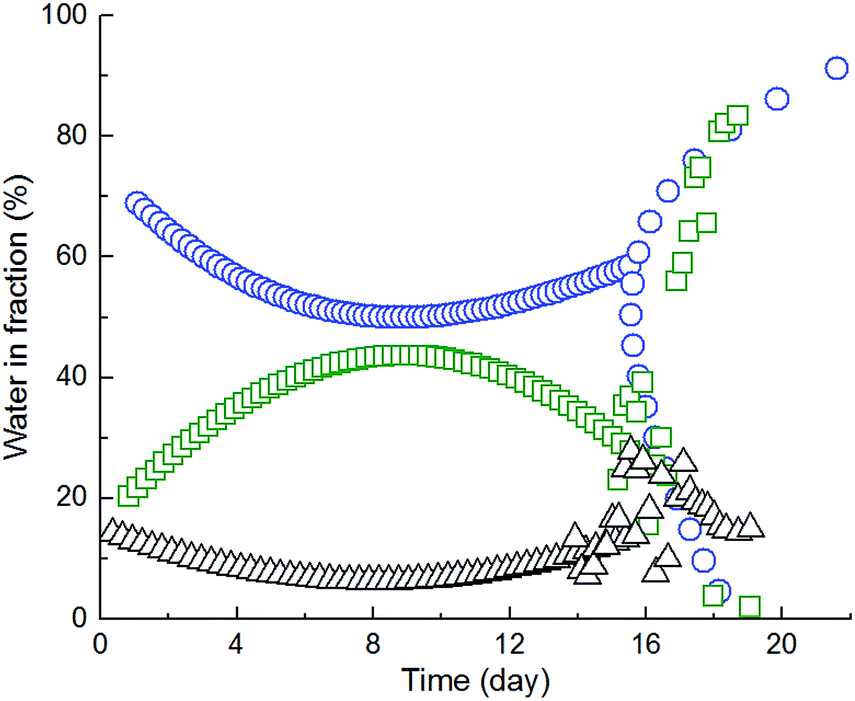 | ||
| Fig. 11 Change in the amount of water fractions in time of sample 63ITAx40, where ○ is amplitudes of free unbonded water, □ amplitudes of weakly bonded water and △ amplitudes of strongly bonded water. | ||
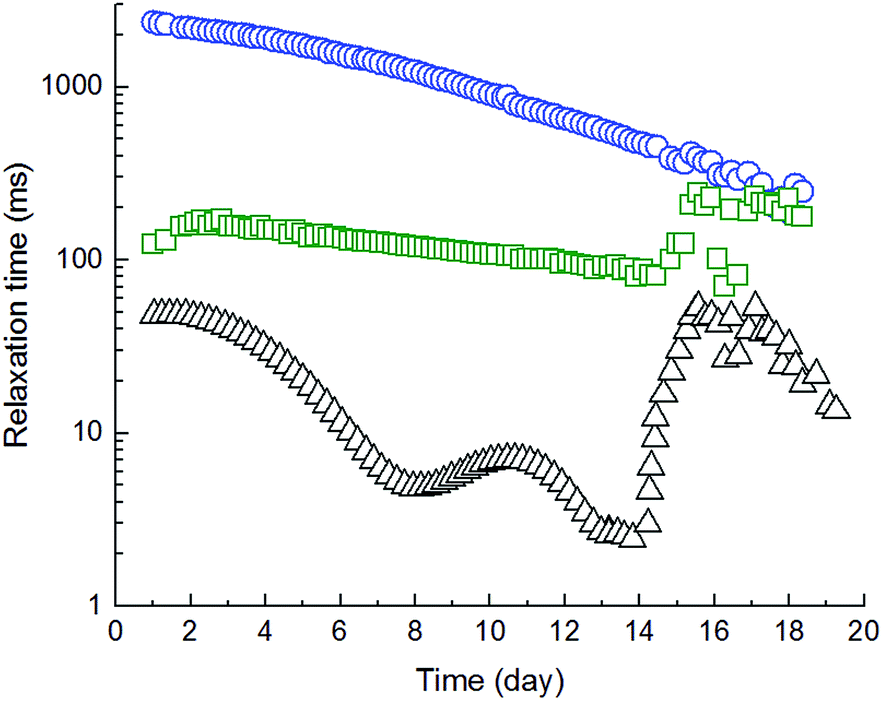 | ||
| Fig. 12 Change in relaxation times of water fractions in sample 63ITAx40 during swelling, where ○ is relaxation time of free unbonded water, □ relaxation time of weakly bonded water and △ relaxation time of strongly bonded water. | ||
Both figures show that the measurement started 24 hours after water addition, when the sample was assumed being already completely wetted and partially swollen. Indeed, the relaxation time reflects the restriction of individual fractions by gel network and it is reciprocally equal to relaxation rate.26 There are two main factors influencing the relaxation: diffusion and the proton exchange between gel-hydroxyl protons and water molecules.26,27
Fig. 12 shows that the highest relaxation time of a water fraction was 2.57 s at the beginning of the experiment and progressively decreased to 200 ms within 18 days. On the contrary, the relaxation time of pure water was determined 2.82 s. Therefore, the decrease in relaxation rate of the slowest fraction (Fig. 11) reflects the progressive incorporation of least affected (bulk) water into the hydrogel structure and the increase in its restriction. The relative amount of this fraction increased till 8th day and then decreased again. The presence of the two other, faster relaxing, water fractions confirms that the gel was already wetted and partially swollen after 24 hours. The relaxation time of these two fractions progressively decreased till 15th day and then suddenly increased. In fact, the amount of the most restricted water fraction (∼10 ms) was constant till 15th day. On the contrary, the amount of the less restricted fraction (∼150 ms) was increasing till 8th day and decreased. Those results imply that gel swelling occurred till 8th day unaffected by other processes. At the 8th day, simultaneously with swelling, started probably the dissolving, which resulted in a structural collapse at 15th day. Degradation of sample was faster than swelling confirming heterogeneous degradation by surface erosion, which is typical for biodegradable polyesters. A small difference in time periods of individual stages of gel swelling and degradation in comparison with Fig. 8 is caused by the experimental arrangement. In fact, during the relaxometry measurement, water was kept constant together with a gel amount at all time and the released lactic and glycolic acids might speed up the gel degradation.
As a result of relaxometry evaluation, the end-linked polyester chains are for certain time hydrolytically stable and water diffusion prevails up to the maximum amount of less restricted water fraction (8th day of 63ITAx40 swelling). Consequently, the amount of free water increased proving the hydrolysis of polymer chains but with slower rate than diffusion which is called bulk erosion. At the day of 15th when the hydrogels exhibited the highest swelling ratio the polymer network undergoes structural collapsing accompanied by the breaking of network nodes followed by chain hydrolysis faster than water diffusion along with sample dissolution proved by GPC (decrease in Mn and broadening PDI). It means that sample undergoes surface erosion. This degradation process is schematically described by Scheme 3 and at Fig. 13. End-linked hydrogel prepared from α,ω-itaconyl-PLGA–PEG–PLGA triblock copolymer (Step I) hydrated in the presence of water (Step II) accompanied by swelling and diffusion of water into the end-linked gel (Step III). Until this time the sample undergo bulk erosion. Increasing time of swelling leads to the degradation (hydrolysis) of ester bonds (Step IV) arising new pores in the hydrogel sample with releasing lactic acid and glycolic acid as degradation products (Step V). The sample follows surface erosion and partially degraded polymer chains are slowly dissolved.
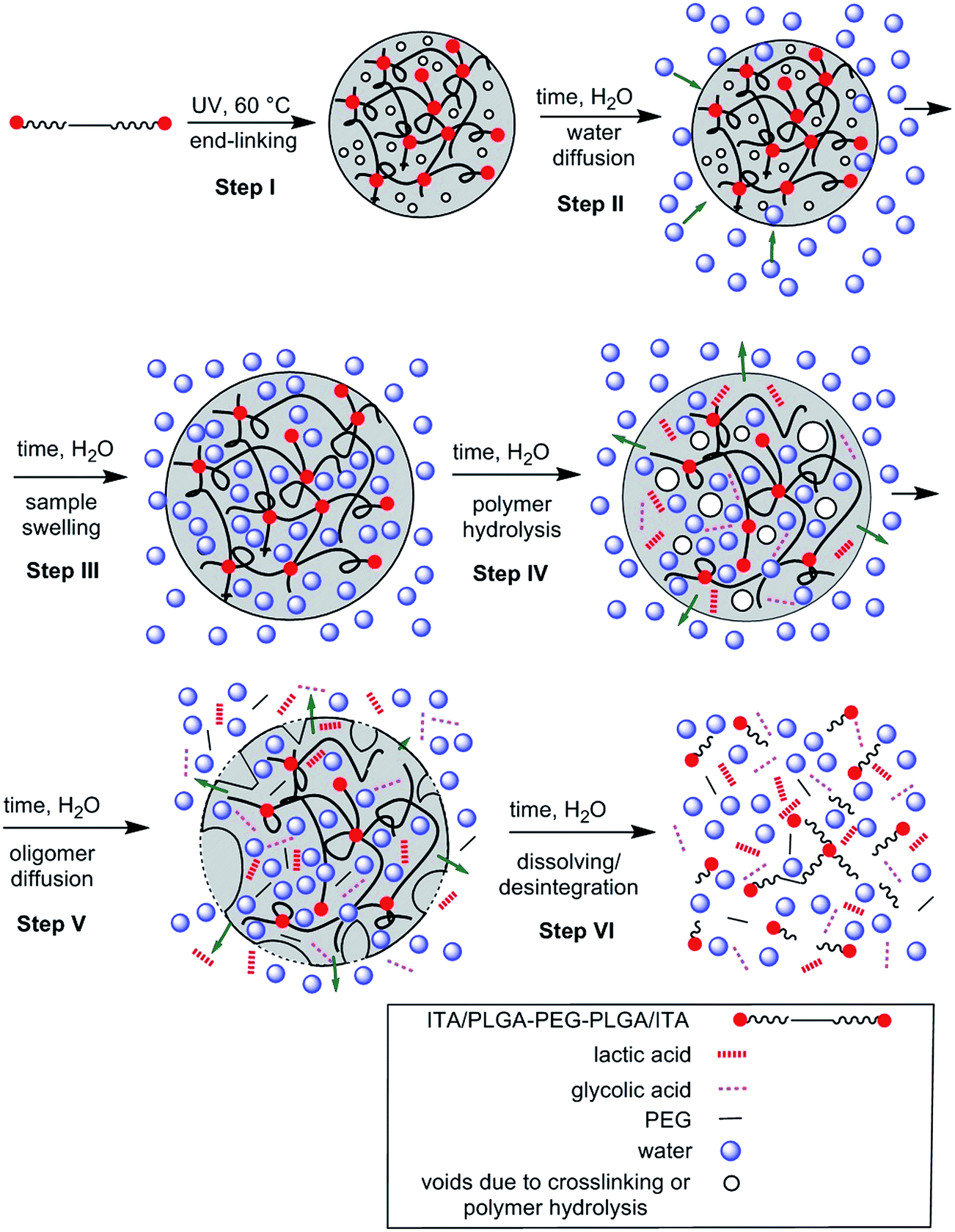 | ||
| Scheme 3 Schema of swelling and degradation of hydrogels. | ||
 | ||
| Fig. 13 Swelling and degradation of hydrogels, (a) end-linked hydrogel, (b) swelled hydrogel, (c) swelled hydrogel with pores, (d) hydrogel with releasing lactic acid and glycolic acid as degradation products. | ||
Conclusions
Here, we showed how to functionalize biodegradable PLGA–PEG–PLGA copolymer with itaconic acid in “one-pot” reaction followed by chemical crosslinking of well-defined α,ω-itaconyl-PLGA–PEG–PLGA macromonomers by light polymerization. Except the reactive double bonds, the itaconic acid brings functional carboxylic groups to both ends of copolymer suitable for further attaching of bioactive compounds (e.g. proteins, drugs). The amount of bonded itaconic acid as well as the crosslinking time enhanced hydrolytic stability and the life-time of end-linked hydrogels. Setting the time of hydrogel degradation is important for optimal adjustment of temporary implants stability in regenerative medicine as well as for controlled drug delivery. Therefore, novel linked hydrogels, made of FDA polymers, can find potential use in moist wound healing or as carriers for controlled drug release.Acknowledgements
This research was carried out under the project CEITEC 2020 (LQ1601) with financial support from the Ministry of Education, Youth and Sports of the Czech Republic under the National Sustainability Programme II. Partial funding under the project 15-18495S from the Czech Grant Agency is greatly appreciated.References
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- L. Yu, H. Zhang and J. Ding, Colloid Polym. Sci., 2010, 288, 1151–1159 CAS.
- C. Pratoomsoot, H. Tanioka, K. Hori, S. Kawasaki, S. Kinoshita, P. J. Tighe, H. Dua, K. M. Shakesheff and F. R. A. J. Rose, Biomaterials, 2008, 29, 272–281 CrossRef CAS PubMed.
- G. M. Zentner, R. Rathi, C. Shih, J. C. McRea, M.-H. Seo, H. Oh, B. G. Rhee, J. Mestecky, Z. Moldoveanu, M. Morgan and S. Weitman, J. Controlled Release, 2001, 72, 203–215 CrossRef CAS PubMed.
- N. L. Elstad and K. D. Fowers, Adv. Drug Delivery Rev., 2009, 61, 785–794 CrossRef CAS PubMed.
- D. Zaldivar, C. Peniche, A. Gallardo and J. Roman, Biomaterials, 1993, 14, 1073–1079 CrossRef CAS PubMed.
- J. S. Temenoff and A. G. Mikos, Biomaterials, 2000, 21, 2405–2412 CrossRef CAS PubMed.
- D. S. Muggli, A. K. Burkoth, S. A. Keyser, H. R. Lee and K. S. Anseth, Macromolecules, 1998, 31, 4120–4125 CrossRef CAS.
- Y.-K. Han, P. G. Edelman and S. J. Huang, J. Macromol. Sci., Part A: Pure Appl. Chem., 1988, 25, 847–869 CrossRef CAS.
- M. P. K. Turunen, H. Korhonen, J. Tuominen and J. V. Seppälä, Polym. Int., 2002, 51, 92–100 CrossRef CAS.
- D. K. Han and J. A. Hubbell, Macromolecules, 1997, 30, 6077–6083 CrossRef CAS.
- A. Helminen, H. Korhonen and J. V. Seppälä, Polymer, 2001, 42, 3345–3353 CrossRef CAS.
- D. Tian, P. Dubois and R. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 2295–2309 CrossRef CAS.
- H. Korhonen, A. Helminen and J. V. Seppälä, Polymer, 2001, 42, 7541–7549 CrossRef CAS.
- M. Ramos, Multi-component Hydrophilic-hydrophobic Systems from Itaconic Anhydride, Doctoral Dissertation, University of Connecticut, 2002 Search PubMed.
- M. Lang and C. C. Chu, J. Appl. Polym. Sci., 2002, 86, 2296–2306 CrossRef CAS.
- J. A. Burdick, L. M. Philpott and K. S. Anseth, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 683–692 CrossRef CAS.
- R. F. Storey, J. S. Wiggins, K. A. Mauritz and A. D. Puckett, Polym. Compos., 1993, 14, 7–16 CrossRef.
- J. Tuominen, J. Kylmä, J. Seppälä and J.-F. Selin, WO 03033563, 2003.
- L. Michlovská, L. Vojtová, L. Mravcová, S. Hermanová, J. Kučerík and J. Jančář, Macromol. Symp., 2010, 295, 119–124 CrossRef.
- Y.-C. Tsai, M.-C. Huang, S.-F. Lin and Y.-C. Su, US 6,171,831, 2001.
- J. P. Loria, M. Rance and A. G. Palmer, J. Am. Chem. Soc., 1999, 121, 2331–2332 CrossRef CAS.
- J. A. Wallach and S. J. Huang, Biomacromolecules, 2000, 1, 174–179 CrossRef CAS PubMed.
- A. J. Ro, S. J. Huang and R. A. Weiss, Polymer, 2008, 49, 422–431 CrossRef CAS.
- P. Conte and G. Alonzo, eMagRes, 2013, 2, 389–398 CAS.
- P. Conte, V. Marsala, C. De Pasquale, S. Bubici, M. Valagussa, A. Pozzi and G. Alonzo, GCB Bioenergy, 2013, 5, 116–121 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Magnification of ATR-FTIR spectrum and thermograph of samples were provided. See DOI: 10.1039/c5ra26222d |
| This journal is © The Royal Society of Chemistry 2016 |