
DFT insights into the cycloisomerization of ω-alkynylfuran catalyzed by planar gold clusters: mechanism and selectivity, as compared to Au(I)-catalysis†
Miao Yangab,
Zhongzhu Chenb,
Yafei Luob,
Jin Zhangb,
Dianyong Tang*b,
Rongxin Hea,
Wei Shena and
Ming Li*a
aSchool of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, P. R. China. E-mail: liming@swu.edu.cn; Tel: +86-23-68366688
bResearch Institute for New Materials Technology and Chongqing Key Laboratory of Environmental Materials and Remediation Technologies, Chongqing University of Arts and Sciences, Chongqing, 402160, P. R. China. E-mail: tangdy2008@163.com; Tel: +86-23-61162836
First published on 22nd February 2016
Abstract
A detailed reaction mechanism of the triatomic gold cluster-catalyzed cycloisomerization of ω-alkynylfuran was systemically investigated via density functional theory at the TPSSh/def2-TZVP level. The computational results indicated that the 5-exo Friedel–Crafts-type mechanism is the most favorable mechanism to form the phenol derivatives. The strong interaction between the gold and vinyl fragments in the Friedel–Crafts adduct is essential for the priority of the 5-exo Friedel–Crafts-type mechanism. Then, the 5-exo Friedel–Crafts-type mechanism on the various planar gold clusters (Au4–10) was studied to clarify the size-effects of the planar gold clusters catalyzed ω-alkynylfuran cycloisomerization. The appropriate interactions between the alkyne group in the substrate and gold clusters play a key role for the 5-exo cyclization step. The energy barriers of the ring-closure of the dienone carbene–gold intermediate step show an interesting “odd–even” behavior respective to the number of gold atoms. The Au3 and Au4 clusters are the most active catalysts for the ω-alkynylfuran cycloisomerization to the phenol derivative. We also found that the active catalyst of the ω-alkynylfuran cycloisomerization catalyzed by the gold(I) complexes should be the gold(0) complexes of the in situ generation. The catalytic activity of the gold(0) complex is comparable with that of the planar gold clusters. These findings may guide the rational design of highly active gold catalysts for the ω-alkynylfuran cycloisomerization to phenol derivatives.
Introduction
Gold-catalyzed phenol synthesis is an isomerization reaction that transforms a ω-alkynylfuran to an annellated phenol via the cleavage of four bonds and the formation of four new bonds. It is an intramolecular reaction of the alkyne–gold complex with the furan ring. After the first report by the Hashmi group in 2000 (Scheme 1),1 many reports about phenol synthesis from ω-alkynylfuran isomerization have emerged over the past few decades (Scheme 1).2–14 Pt(II), Pd(II), Ir(I), and Rh(I) were also found to catalyze the intramolecular reactions of alkynes with arenes and furans (Scheme 2).2,13,14 Still, gold complexes are clearly more active than other metals, display effective chemoselectivity and high synthetic efficiency in the cycloisomerization of ω-alkynylfurans, and yield no significant side products under mild conditions (Scheme 1).2,3,9 The cationic binuclear gold(I) complex [(Ph3PAu)2Cl]BF4 could catalyze the cyclization of ω-alkynylfurans to the main product phenols with the annellated furans as the side-products.4 The 1,2,3-triazole was used as a special “X-factor” (as a ligand for [AuPPh3]+ coordination forming a [Ph3P–Au–triazole]+ complex) to stabilize the gold catalyst and obtain excellent yields and chemoselectivity by Shi group.9 The X-factor, which was developed by using the 1,2,3-triazole as a coordination factor to stabilize the catalyst, could be regarded as an alternative strategy, instead of tuning the phosphorus ligands.9 The Hashmi group also found that the mononuclear NAC–gold(I) and phosphite–gold(I) catalysts are highly active for the cyclization of ω-alkynylfurans. Turn-over numbers up to 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for the cyclization of ω-alkynylfurans were obtained.11,12 Gold nanoparticles supported on nanocrystalline CeO2 could also catalyze the cycloisomerization of ω-alkynylfurans to phenols.15
000 for the cyclization of ω-alkynylfurans were obtained.11,12 Gold nanoparticles supported on nanocrystalline CeO2 could also catalyze the cycloisomerization of ω-alkynylfurans to phenols.15
 | ||
| Scheme 1 Gold-catalyzed cycloisomerizations of ω-alkynylfurans. | ||
 | ||
| Scheme 2 Different transition metal complex-catalyzed-ω-alkynylfurans cycloisomerizations. | ||
Remarkable works have also been performed regarding the elucidation of this mechanism including the successful isolation of several key intermediates, that is, the arene oxide and oxepine.7,16 The breakage and formation of four bonds during this reaction is clearly not an elementary reaction. Because the rate-limiting step possesses lowest reaction rate in the overall reactions (reaction kinetic theory), the reaction intermediate of this step should be opulent and be detectable by the NMR, IR etc. Therefore, based on the kinetic theory and 1H NMR spectroscopy, Hashmi et al. concluded that the failure to detect intermediates with AuCl3 simply means that the first step was the rate-limiting step.17 Hashmi et al. demonstrated that the oxygen atom is transferred intramolecularly during the reaction.2 The Hashmi group showed that the reaction does not proceed via an alkynyl or a vinylidene complex because a primary kinetic isotope effect was not observed with a substrate deuterated at the alkyne.16 The Echavarren group found that the intramolecular reaction of furans with alkynes catalyzed by PtCl2 is mechanistically related to that of enyne in polar solvents.13,14 The PtCl2-catalyzed reaction is initiated by the nucleophilic attack of the furan on a (η2–alkyne)–Pt(II) complex to form a cyclopropyl platinum carbene via density functional theory calculations. According to 18O labeling experiments,1 substrate deuteration studies,16 and in situ NMR spectroscopy,7,17 Hashmi et al. proposed a mechanism for the Au(III)-catalyzed phenol synthesis reaction shown in Scheme 3. The reaction kinetic studies provided strong evidence that the actual Au(I) catalyst was likely the Ph3PAu+ complex, which was under equilibrium with precatalyst [Ph3P–Au–triazole]+ as shown by the Shi group.9 Recently, Oliver-Meseguer et al. used matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry (MALDI-TOF) and ultraviolet-visible (UV-vis) spectroscopy to report that very small (3–6 atoms) gold clusters generated in situ from gold salts, complexes, and/or nanoparticles can catalyze this reaction.18 Moreover, the small gold clusters (only 3 to 10) can be formed from different Au(I) and Au(III) salts or gold complexes during a catalytic process in solution at room temperature in some cases; they can efficiently activate the C–C triple bond of alkynes.19–21 For instance, the ester-assisted hydration of alkynes catalyzed by small gold clusters could yield an unprecedented ∼107 turnover number (TON).19 In addition, the ligand can play some significant roles in the activity of the catalyst, including two main aspects. One respect is the gold catalysts in different oxide state and ligands can cause different product distributions. As shown in Scheme 1, gold catalysts in different oxidation states and ligand produce different product distributions.1–12 Another aspect is the ligand can exert a straightforward influence on the formation and stability of the small clusters, which has been demonstrated by kinetic and spectroscopic studies.18–21 Distinctly, the formations of gold clusters and the catalytic activity inevitability depend on the chemical nature of ligands and counter anions in the complexes, which is reported by Corma group.21 Meanwhile, the gold complexes with different ligands can cause obviously catalytic activity, which can be found in many previous studies.1–12,21
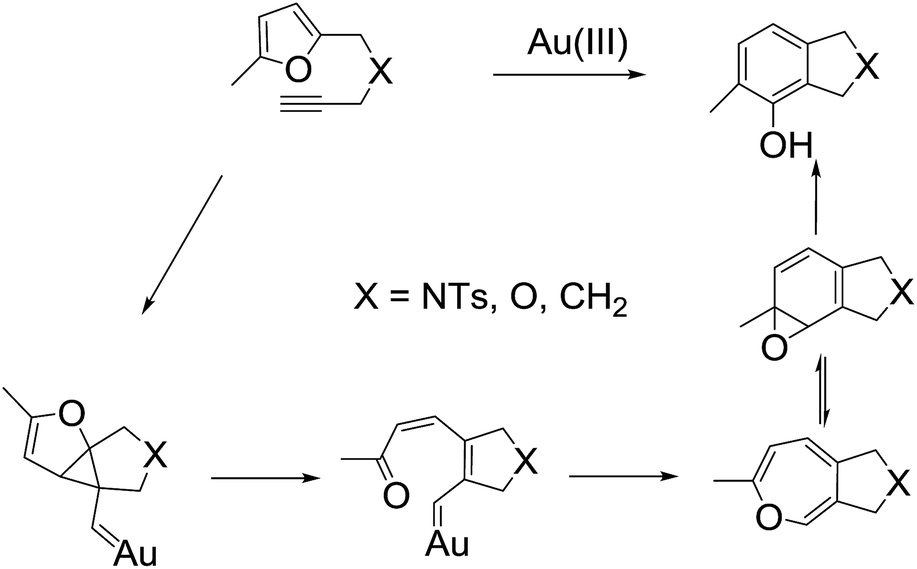 | ||
| Scheme 3 Proposed mechanism for the gold-catalyzed phenol synthesis. | ||
The previous experimental studies also showed that the formation of phenol should proceed via arene oxide or oxepine intermediate because of their capture in a Diels–Alder reaction with dienophiles and detection with in situ NMR spectroscopy.7,17 However, the formation of arene oxide or oxepine intermediate from reactants under experimental conditions remains unclear. Other questions include: which gold species is the active catalyst in these reactions? How do we control the product selectivity via active gold species? To answer some of these questions, the planar gold clusters were chosen as the active catalysts to investigate the detailed mechanism of the ω-alkynylfurans cycloisomerization. The planar gold clusters were assumed to be the active catalyst of the ω-alkynylfuran cycloisomerization catalyzed by Au(I) and Au(III) complexes in some cases.18,21 In our previous papers, the alkyne activation reactions including the cycloisomerization of 1,6-enyne and the cycloisomerization/oxidative dimerization of phenyl propargyl ether catalyzed by the Au38 cluster were investigated.22,23 The present theoretical mechanistic investigations are aimed at extending the understanding of ω-alkynylfuran cycloisomerization catalyzed by the planar gold clusters by elucidating the following intriguing, but not yet firmly resolved aspects: (1) which is the preferred mechanism for the ω-alkynylfuran cycloisomerization catalyzed by the planar gold clusters? (2) What is the source of product selectivity? (3) How does the size of the planar gold clusters affect the catalytic activity? (4) Which factor controls the catalytic activity of the planar gold clusters? (5) Which is true active catalyst for the ω-alkynylfurans cycloisomerization catalyzed by the gold(I) complexes? It should be noted that our efforts focused on the mechanism of ω-alkynylfuran cycloisomerization catalyzed by the planar gold clusters and AuPPh3+ complex. The truly active catalyst and mechanism of the ω-alkynylfuran cycloisomerization catalyzed by the different oxidation state gold species (such as AuCl, AuCl3, Au(I) complexes, and Au/CeO2) will be investigated in the future.
Computational details
The mechanism was investigated via density functional theory (DFT) using the TPSSh functional24,25 with the def2-TZVP26 basis sets as implemented in Gaussian 09 package,27 which was used in the geometric optimizations of intermediates (IMs) and transition states (TSs). To check the IMs and TSs structures, vibrational frequency calculations at the same level of theory were performed. Intrinsic reaction coordinates (IRC)28,29 were performed to confirm the transition states connecting with the corresponding reactant and product intermediates. According to reaction conditions, the solvent effect of acetonitrile (ε = 37.5) was evaluated using the SMD model (where “SM” and “D” stand for solvation model and density, respectively).30 Natural charges were calculated via natural population analysis at the same level as that used for geometry optimization.The adsorption energies of the gold cluster substrates are defined in eqn (1).
| ΔEad = Etotal − Ecluster − Esubstrate | (1) |
It should be noted that that the spin–orbital coupling can play important role in the calculation of both, the atomization energies and the relative stability of the isomers of gold clusters.31 However, due to the large computational times, the spin–orbital coupling is not considered in the gold-catalyzed reactions, such as CO oxidation, C–C coupling reaction, water–gas shift reaction, et al.32–36 Similarly, the spin–orbital coupling is not considered in the current paper.
Results and discussion
It should be noted that the charge states of the gold clusters are neutral in the present study. The reasons are mainly collected as the following two points. Firstly, according to the experimental results, the detection of in situ UV-vis characteristic peek shows the formation of neutral Au3 and Au4 clusters,18–21 not the Aun+ clusters. Secondly, we also calculate the reaction mechanisms via using the Au3+ and Au4+ clusters as the catalysts. However, the TS2/3 and TS3/4 could not be located with our best endeavor. The potential energy surface scan unveils that the energy is increasing for IM2 → IM3 and IM3 → IM4. Therefore, in order to consistent with the experimental results,18–21 the Aun+ clusters are not considered as the active catalysts in this manuscript.In the present paper, the hybrid meta-GGA functional TPSSh was utilized because of the very good metal–ligand bond energies, cohesive energy of metal clusters, and structural parameters of metal clusters it is reported to provide.37–42 To confirm our choice of functional and basis set, the possible structures for Au3–10 were optimized and shown in Fig. S1.† The predicted bond lengths of the Au–Au bond in Au2 and Au3 is 2.518 and 2.563 Å, which is in line with those at the CCSD(T)-F12a/def2-TZVP levels (2.476 and 2.519 Å), respectively.43 The cohesive energies of Au2 and Au3 are 24.32 and 24.63 kcal mol−1, which is also in agreement with the experimental results (26.82 and 29.23 kcal mol−1), respectively.44–46 For the Au3 cluster, the acute triangle structure is higher than the obtuse triangle structure about 1.67 kcal mol−1, which is in line with results obtained at the various functionals and basis sets.31,47 As shown in Fig. S1,† the stable gold clusters of each gold cluster are in line with the previous results.31,43,47–49 Therefore, the functional and basis sets could produce the reliable results for the structural and properties of the gold clusters.
Mechanism for ω-alkynylfuran cycloisomerization catalyzed by Au3 cluster
To illustrate the catalytic behavior of planar gold clusters, the Au3 cluster was selected as a model catalyst to simulate the detailed mechanism of the ω-alkynylfuran cycloisomerization. The Au3 cluster was assumed to be the active catalyst of the ω-alkynylfuran cycloisomerization catalyzed by Au(I) and Au(III) complexes in some cases.18 According to the mechanism proposed by the Hashmi group,7,15–18 the possible pathways of the ω-alkynylfuran cycloisomerization catalyzed by Au3 clusters were designed and shown in Scheme 4. The energy profiles, optimized structures, and related parameters are depicted in Fig. 1–5.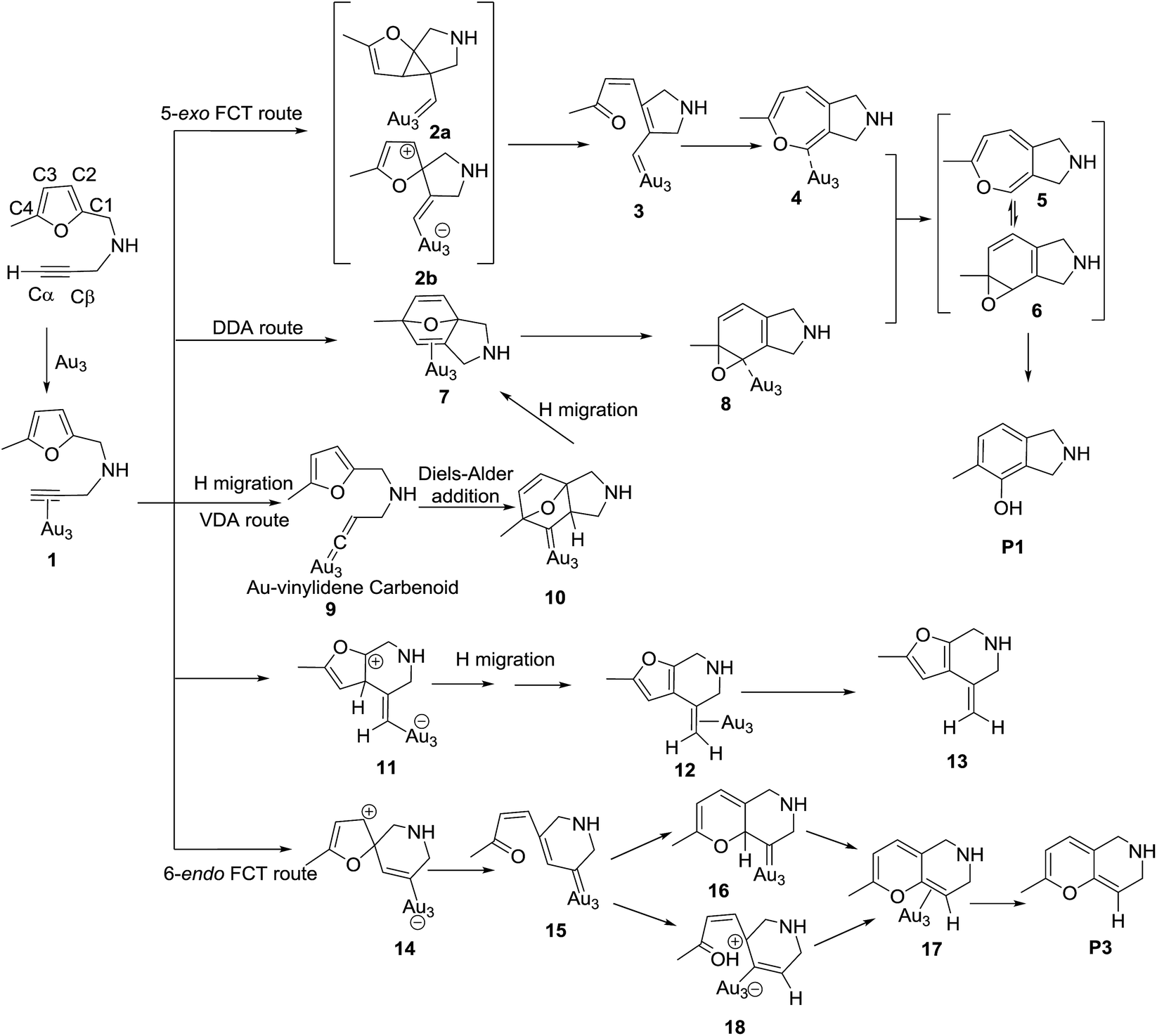 | ||
| Scheme 4 The 5-exo Friedel–Crafts-type (FCT) route, direct Diels–Alder-type (DDA) route, and Au–vinylidene carbenoid (VDA) route to form the phenol derivative (P1), the formation of β-alkenylated furan (P2), and the 6-endo Friedel–Crafts-type (FCT) route toward the pyran derivative (P3). | ||
 | ||
| Fig. 1 The optimized structures, related parameters, potential energy profile, and relative Gibbs free energies (colored by red, 298.15 K and 1 atm) of the 5-exo FCT pathway to form the phenol derivative P1 in the gas phase and acetonitrile solvent (parentheses) are shown. The bond length is in Å. | ||
Adsorption of ω-alkynylfuran
It is well-known that the geometric structure of neutral Au3 cluster is obtuse triangle.31,47,50 As shown in Fig. 1, the adsorption of the ω-alkynylfuran on the Au3 cluster induces the structure deformation of the Au3 cluster into acute triangle (Fig. 1). The adsorption energy of the substrate on the Au3 cluster is −32.84 (−29.05) kcal mol−1 in the gas phase due to the strong σ-donation from the π orbital of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond as well as the partially occupied d orbital of the gold atom and the π-back-donation from the d orbital of the gold atom to the π* orbital of the CC bond. The interaction between the alkyne fragment and Au3 is similar to that of alkyne fragments of 1,6-enyne and aryl propargyl ether with the surface gold atom of the Au38 clusters.22,23 The adsorption of substrate causes the net transfer of electron from Au3 fragment to substrate fragment (the natural charge on the Au3 fragment is about 0.17|e|). The received electron from the substrate fragment is mainly distributed on the CC bond, which enhances the nucleophilicity of two carbon atoms. Hence, the adsorption of substrate favors the subsequent nucleophilic addition. At the same time, the adsorption of substrate results in the activation of CC bond, which is reflected by the increasing CC bond length from 1.20 Å to 1.25 Å due to the back-donation of d electrons to the π* orbital of the CC bond.
C bond as well as the partially occupied d orbital of the gold atom and the π-back-donation from the d orbital of the gold atom to the π* orbital of the CC bond. The interaction between the alkyne fragment and Au3 is similar to that of alkyne fragments of 1,6-enyne and aryl propargyl ether with the surface gold atom of the Au38 clusters.22,23 The adsorption of substrate causes the net transfer of electron from Au3 fragment to substrate fragment (the natural charge on the Au3 fragment is about 0.17|e|). The received electron from the substrate fragment is mainly distributed on the CC bond, which enhances the nucleophilicity of two carbon atoms. Hence, the adsorption of substrate favors the subsequent nucleophilic addition. At the same time, the adsorption of substrate results in the activation of CC bond, which is reflected by the increasing CC bond length from 1.20 Å to 1.25 Å due to the back-donation of d electrons to the π* orbital of the CC bond.
Pathways for the formation of phenol
As shown in Scheme 4, after the adsorption of substrate on Au3 cluster, there are four possible pathways to produce phenol derivatives. The first one is 5-exo Friedel–Crafts-type reaction route starting from the nucleophilic addition of a Cβ atom to the C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2 bond or C1 site of furan ring (abbreviated as 5-exo FCT). The second pathway is the [4 + 2] cycloaddition between the CC bond and the C1C2–C3C4 of the furan ring to produce the final product (abbreviated as DDA). The last pathway is a direct hydrogen transfer from the Cα atom to the Cβ atom to form an Au–vinylidene carbenoid intermediate, and subsequent Diels–Alder addition of the Cα–Cβ double bond to the furan ring (abbreviated as VDA). It is unlikely that the pathway proceeds via an alkynyl or a vinylidene complex because a primary kinetic isotope effect was not observed with a substrate deuterated at the alkyne,16 but this pathway is presented for comparison. In the following sections, the 5-exo FCT, DDA, and VDA pathways are discussed in further detail.
C2 bond or C1 site of furan ring (abbreviated as 5-exo FCT). The second pathway is the [4 + 2] cycloaddition between the CC bond and the C1C2–C3C4 of the furan ring to produce the final product (abbreviated as DDA). The last pathway is a direct hydrogen transfer from the Cα atom to the Cβ atom to form an Au–vinylidene carbenoid intermediate, and subsequent Diels–Alder addition of the Cα–Cβ double bond to the furan ring (abbreviated as VDA). It is unlikely that the pathway proceeds via an alkynyl or a vinylidene complex because a primary kinetic isotope effect was not observed with a substrate deuterated at the alkyne,16 but this pathway is presented for comparison. In the following sections, the 5-exo FCT, DDA, and VDA pathways are discussed in further detail.
Firstly, the 5-exo FCT reaction route is discussed (Fig. 1). After formation of IM1, the nucleophilic addition of the Cβ atom to the C1 site of the furan ring results in a metastable spiro-intermediate IM2 (the 5-exo cyclization step). Because the delocalization of the negative charge in IM2 is not sufficient to compensate for the stability of the gold vinyl, this step is energetically unfavorable and is endothermic by 6.86 and 1.04 kcal mol−1 in the gas phase and acetonitrile solvent, respectively. This step needs to overcome about 19.06 and 15.87 kcal mol−1 in the gas phase and acetonitrile solvent, respectively. We also considered the nucleophilic addition of Cβ atom to the C1–C2 bond of furan to give the gold–cyclopropylcarbene complex (2a, Scheme 4), but only a minimum-energy structure could be located. The transition state connecting with the IM1 and gold–cyclopropylcarbene complex could not be located. The predicted energy of the gold–cyclopropylcarbene complex is higher than that of IM2 by about 5 kcal mol−1, which indicated that the formation of the gold–cyclopropylcarbene complex is thermodynamically unfavorable.
This point is significantly different from the PtCl2-catalyzed intramolecular reaction of furan with alkyne in which the formation of the Pt–cyclopropylcarbene complex is thermodynamically and kinetically favorable.13,14 The charges on Cβ, C1, and C2 atoms of IM1 are −0.10, 0.27, and −0.31|e|, respectively, which supports the formation of the Friedel–Crafts complex IM2. The C1–O bond is lengthened to 1.48 Å in IM2 indicating the activation of a C–O bond.
This activation is favorable for the following step of the C1–O bond cleavage. Subsequently, the cleavage of the C1–O bond forms a conjugate dienone carbene–gold intermediate IM3 (the ring-opening of furan step). IM3 should be more stable than IM2 due to the strong delocalization of charges in IM3. As expected, this step is exothermic by about 9.80 and 13.31 kcal mol−1 in the gas phase and acetonitrile solvent, respectively. The energy barrier of the C1–O bond cleavage is only 8.48 and 8.44 kcal mol−1 in the gas phase and acetonitrile solvent, respectively, because of the strong activation of the C1–O bond in IM2.
Next, the oxygen atom of the C4O bond attacks on the Au–Cα bond to form a gold–oxepine complex IM4 (the ring-closing of the dienone carbene–gold intermediate). This step is exothermic by about 10.35 and 1.05 kcal mol−1 with energy barriers of 12.22 and 16.99 kcal mol−1 in the gas phase and acetonitrile solvent, respectively. Finally, the oxepin derivative IM5 is desorbed from the Au3 cluster, which is endothermic by about 31.35 and 27.99 kcal mol−1 in the gas phase and acetonitrile, respectively. The benzene oxide IM6 and the oxepin derivative IM5 are in a tautomeric equilibrium during the reaction with an energy barrier of 5.42 (4.64) kcal mol−1 in the gas phase (acetonitrile). Finally, IM6 is quickly converted into product phenols with an exothermicity of 39.44 (40.21) kcal mol−1 with heating or acid.1,2,17 Of note, the benzene oxide IM6 was captured via a Diels–Alder reaction with dienophiles.17 In summary, the rate-limiting step of the 5-exo FCT reaction route is the nucleophilic addition of the Cβ atom to the C1 site of the furan ring to form the spiro-intermediate IM2 with an energy barrier of 19.06 kcal mol−1 in the gas phase—the formation of the gold–oxepine complex IM4 is the rate-determining step in acetonitrile.
In order to further prove the reasonability of the functional and basis set used in this paper, the 5-exo FCT pathway was calculated at the PBE/def2-TZVP levels. The calculated results are shown and summarized in Fig. S2 and Table S1.† As shown in Fig. 1 and S2,† the geometric parameters of the related structures with the TPSSh and PBE functionals are very similar. The adsorption energies of substrate, and energy barriers, and the reaction energies at the TPSSh/def2-TZVP and PBE/def2-TZVP levels are similar. Therefore, results with the TPSSh functional and def2-TZVP are reliable for the gold cluster-catalyzed the ω-alkynylfuran cycloisomerization.
We next focus on the pathway initializing from the [4 + 2] cycloaddition between the CαCβ bond and C1C2–C3C4 of furan ring in IM1, i.e. the DDA pathway (Fig. 2 and 3). The Diels–Alder adduct IM7 (Fig. 2) has higher energy than IM1 (2.54/2.12 kcal mol−1 in the gas phase/acetonitrile). The corresponding transition state TS1/7 must overcome an energy barrier of 25.32 (26.24) kcal mol−1 in the gas phase (acetonitrile). The Cα and Cβ atoms in IM7 are approximately pyramidal, and the structure resembles a η2–gold–ene complex. Next, the bridged O atom would shift to the Cα atom in IM7 along with the breakage of the C1–O bond to form a gold–benzene oxide complex IM8. This step must overcome an energy barrier of 32.44 (25.44) kcal mol−1 in the gas phase (acetonitrile). Due to migration of the double bond from the Cα–Cβ bond to the C1–Cβ bond in the process of IM7 → IM8, the interaction between the Au3 cluster and the benzene oxide is very weak as reflected by the bond length of the Au–Cα and Au–Cβ bonds (Fig. 3). The weak interaction also results in the endothermicity of IM7 → IM8. Finally, the decomposition of IM8 gives a Au3 cluster and benzene oxide with slight endothermicity. Clearly, the migration of the bridged O atom step (IM7 → IM8) is the rate-limiting step for the DDA pathway with an energy barrier of 32.44 (25.44) kcal mol−1 in the gas phase (acetonitrile).
 | ||
| Fig. 2 Potential energy profiles and relative Gibbs free energies (colored by red, 298.15 K and 1 atm) of the DDA and VDA pathways to form phenol derivative P1 in the gas phase and acetonitrile solvent (parentheses). The bond length is in Å. | ||
 | ||
| Fig. 3 The optimized structures and related parameters for DDA and VDA pathways (bond length in Å). | ||
Finally, the pathway via the Au–vinylidene carbenoid complex (VDA pathway) (Fig. 2 and 3) was investigated. The gold–vinylidene carbenoid complexes are highly reactive intermediates—they are the key intermediate for many gold-catalyzed organic reactions.51–57 The formation of Au–vinylidene carbenoid intermediate IM9 is endothermic by about 10.06 (7.98) kcal mol−1 with an energy barrier of 44.95 (41.12) in the gas phase (acetonitrile). Obviously, the formation of the Au–vinylidene carbenoid intermediate IM9 is unfavorable thermodynamically and kinetically, which agrees with the experimental findings.16 After the formation of the Au–vinylidene carbenoid intermediate IM9, the Diels–Alder adduct IM10 is produced with an energy barrier of 28.43 (30.20) kcal mol−1 in the gas phase (acetonitrile). Due to the high ring tension, the Diels–Alder process (IM9 → IM10) is endothermic. Subsequently, the hydrogen atom on the Cβ atom transfers to the Cα atom in IM10 to give rise to IM7. The activation energy is predicted to be 29.79 (29.65) kcal mol−1 in the gas phase (acetonitrile). The remaining steps toward benzene oxide IM6 are the same as that of the DDA pathway. The rate-limiting step of the VDA pathway is the formation of a Au–vinylidene carbenoid intermediate via the hydrogen transfer with an activation energy of 44.95 (41.12) in the gas phase (acetonitrile). Hence, this step is also unfavorable kinetically.
In summary, these findings indicate that the ω-alkynylfuran cycloisomerization to phenol catalyzed by a Au3 cluster mainly proceeds via the 5-exo FCT pathway. All stationary points for the 5-exo FCT pathway stand below the separated catalyst and reactant, hence, the 5-exo FCT pathway proceeds easily. The rate-limiting step of the 5-exo FCT pathway is the first step, i.e. the 5-exo nucleophilic addition of Cβ atom to furan ring (IM1 → IM2). This point is highlighted by the fact that no intermediate could be detected in the experiments, which concurs with the experimental data.17 The formation of Diels–Alder adduct IM7 and Au–vinylidene carbenoid intermediate IM9 are both thermodynamically and kinetically unfavorable.
Pathways for the formation of pyran derivative
There is a 6-endo FCT pathways, i.e., the nucleophilic addition of a Cα atom to the furan ring in IM1, which is in competition with the 5-exo FCT pathway. The 6-endo FCT pathway finally leads to a pyran derivative. The optimized structures, related parameters, and potential energy profiles are shown in Fig. 4 and 5. | ||
| Fig. 4 Potential energy profiles and relative Gibbs free energies (colored by red, 298.15 K and 1 atm) of the 6-endo FCT pathway toward pyran derivative P3 in the gas phase and acetonitrile solvent (parentheses). | ||
 | ||
| Fig. 5 The optimized structures and related parameters for the 6-endo FCT pathway toward pyran derivative P3 (bond length in Å). | ||
The 6-endo nucleophilic addition of Cα atom to the C1 atom of the furan ring in IM1 results in a metastable spiro intermediate IM11 via TS1/11. The predicted activation energy is 20.19 (18.94) kcal mol−1 in the gas phase (acetonitrile), which is slightly higher than that of formation in the 5-exo nucleophilic addition intermediate IM2. Subsequently, cleavage of the C1–O bond of the furan ring in IM11 produces intermediate IM12—the C4–O single bond changes to a C4–O double bond. This process requires an energy barrier of 7.29 (7.20) kcal mol−1 with an exothermicity of 8.16 (12.53) kcal mol−1 in the gas phase (acetonitrile).
Next, there are two pathways from IM12 to the final product P3. The first pathway is via the O atom of the C4O bond that attacks the Cα atom to result in the six-membered ring intermediate IM13. Then, the H atom on the Cα atom migrates to the Cβ atom to generate product complex IM14. The corresponding energy barriers are 23.97 (29.26) (IM12 → IM13) and 6.37 (6.88) (IM13 → IM14) kcal mol−1 in the gas phase (acetonitrile).
Alternately, the migration of the H atom from the Cα atom to the Cβ atom overcomes an energy barrier of 55.48 (60.56) kcal mol−1 to produce intermediate IM15. Subsequently, the carbonyl group nucleophilicly attacks the Cα atom to result in product complex IM14. The activation energy is only 9.76 (17.65) kcal mol−1 (TS15/14) with an exothermicity of 27.89 (17.91) kcal mol−1. Comparison of the two hydrogen steps (IM12 → IM15 and IM13 → IM14) shows that the higher energy barriers of IM12 → IM15 are ascribed to the strong C(sp2)–H bond in IM12. Simultaneously, the electron-deficient Cα atom in IM15 results in a low energy barrier of TS15/14. Finally, the pyran derivative P3 is desorbed from IM14 with regeneration of Au3 cluster, which is endothermic by 31.21 (28.08) kcal mol−1 in the gas phase (acetonitrile). Obviously, the energy barriers of formation for the pyran derivative are higher than that of the 5-exo FCT pathway.
The pathway for the formation of the β-alkenylated furan (P2) has also been studied. However, only the minimum of 11 was located. The transition state connecting IM1 and 11 could not be obtained with our best efforts. At the same time, the transition states of the Cβ migration from the C1 atom to the C2 atom (IM2 → 11) as well as the gold–cyclopropylcarbene complex to 11 could not be located. The charges on the C1 and C2 atoms of the furan ring in IM1 are 0.23 and −0.29|e|, respectively. Hence, the Cα atom could preferably attack the C1 atom to form IM2. Therefore, the formation of β-alkenylated furan is not favorable for the ω-alkynylfuran cycloisomerization catalyzed by Au3 cluster.
To better understand the 5-exo FCT pathway, DDA pathway, and 6-endo FCT pathway, we performed energy decomposition analysis (EDA)22,23,58–60 on the energy barriers and reaction energy for the first steps of the three pathways (IM1 → IM2, IM1→ IM7, and IM1 → IM11). The details of the EDA were supplied in the ESI.† Concisely, the energy barriers and reaction energies are decomposed into three parts. The first part is the deformation (distortion) energies of substrate fragment (Sub) from reactants to transition states (products). The second part is the deformation (distortion) energies of catalyst fragment (Au3, Ca) from reactants to transition states (products). The last part is the change in interaction energy (ΔEb) between the Sub and Ca fragments from reactants to transition states or products. The EDA results are shown in Table 1 and indicate that the deformation energies of the Au3 fragments for these processes are very small suggesting only small structural changes in the Au3 fragments. The changes in binding energies are slightly favorable for the three pathways. The energy barriers are mainly due to the deformation of the Sub fragments. Simultaneously, the differences in energy barriers of the three pathways originate from the structural deformation of a Sub fragment. The changes in binding energies between Au3 and Sub fragments counteract the deformation energies of the Sub fragments in IM2 and IM7. Therefore, the strong interaction between gold and vinyl in Friedel–Crafts adduct stabilizes the Friedel–Crafts addition intermediates. This results in high selectivity toward the phenol derivative.
| Reactions | ΔEint-1 | ΔEint-2 | ΔEb | ΔED-Sub | ΔED-Ca | ΔrEa/ΔrE |
|---|---|---|---|---|---|---|
| IM1 → TS1/2 | −45.92 | −49.49 | −3.57 | 23.20 | −0.04 | 19.59 |
| IM1 → TS1/7 | −45.92 | −50.36 | −4.44 | 29.93 | 0.07 | 25.56 |
| IM1 → TS1/11 | −45.92 | −48.20 | −2.28 | 22.95 | 0.02 | 20.69 |
| IM1 → IM2 | −45.92 | −82.84 | −36.92 | 44.31 | −0.64 | 6.75 |
| IM1 → IM7 | −45.92 | −51.26 | −5.34 | 6.06 | 0.07 | 0.79 |
| IM1 → IM11 | −45.92 | −76.68 | −30.76 | 35.26 | −0.65 | 3.85 |
Size-effect of planar gold clusters
Oliver-Meseguer et al. found the Au3–10 clusters were formed in reaction progress. Simultaneously, the various-sized gold clusters showed different catalytic activity for alkyne activation reactions.18–21 Therefore, the mechanism on Au4–10 clusters were investigated to illustrate the catalytic activity of the planar gold clusters toward the ω-alkynylfuran cycloisomerization. Because our goal is to explore the reaction activity of neutral planar gold clusters, compared with that of Au3 and Au4 clusters. Therefore, although many reports show the structures (both 2D and 3D) of bigger clusters are closely related to the calculation methods,49,61,62 the relatively stable planar structures of Au5–10 clusters are chosen in the present article (Fig. 6). At the same time, as shown in Fig. S1,† the most stable structures of Au3–10 are planar at the TPSSh/def2-TZVP level. From above discussions on Au3 cluster-catalyzed ω-alkynylfuran cycloisomerization, the 5-exo FCT mechanism was the most favorable mechanism to synthetize phenol. So, only the 5-exo FCT mechanism on Au4–10 clusters were studied. The energy profiles, optimized structures, and related parameters are shown in Fig. S3–S9.† The TS3/4 of Au6 cluster was not located in this study. The relaxed scan along the Cα–O bond (Fig. S10†) showed no energetic maximum between IM3 and IM4 for Au6 cluster. The relaxed scan of potential energy profile for TS3/4 in the Au8 cluster was also performed (Fig. S10†). | ||
| Fig. 6 The structures and isosurfaces (cutoff = 0.03) of the HOMO and LUMO orbitals of the planar gold clusters. | ||
Firstly, the adsorption of ω-alkynylfuran on these clusters was analyzed. The adsorption energy of ω-alkynylfuran on the Au4 cluster is −33.58 and −29.64 kcal mol−1 in the gas phase and acetonitrile solvent, respectively. This is comparable to that of the Au3 cluster (−32.84 and −29.05 kcal mol−1). It is interesting that the ω-alkynylfuran is preferentially adsorbed on the acute vertex of the Au4 cluster. To confirm this point, the isosurface of the highest occupied molecular orbital (HOMO) and lowest occupied molecular orbital (LUMO) of the Au4 cluster were drawn (Fig. 6). Fig. 6 highlights that the main contribution of the LUMO is the acute vertex of the Au4 cluster, which agrees with the adsorption of the substrate.
The adsorption energies of the ω-alkynylfuran on the Au5–10 clusters are in the range of −21 to −16 kcal mol−1 (Table 2). These results suggest that ω-alkynylfuran is more favorably adsorbed on the Au3 and Au4 clusters than on other planar gold clusters (Au5–Au10). The higher adsorption energies on the Au3 and Au4 clusters could be attributed to the large back-donation from the d orbital of the gold atom to the π* orbital of the CαCβ bond. The different back-donation could be characterized by the different bond lengths of the CαCβ bond. The binding strength of the substrate with gold is controlled by the degree of charge transfer and two different electron density transfer processes: σ-donation from the alkyne fragment to the gold and π-back-donation from the gold to the alkyne fragment. Therefore, the strength of the interaction depends on two factors: the difference in energy and the spatial overlap between the orbitals involved. The HOMO and LUMO orbitals of planar gold clusters consist of several lobes (Fig. 6). These are mainly localized on the edge and corner gold atoms and are fully accessible to interaction with the alkyne fragments of the substrate. This efficient overlap between the molecular orbitals explains the higher adsorption energy values and the preferred adsorption sites.
| GC | ΔrEad | ΔrEa (TS1/2) | ΔrEa (TS2/3) | ΔrEa (TS3/4) |
|---|---|---|---|---|
| Au3 | −32.84 (−29.05) | 19.06 (15.87) | 8.48 (8.44) | 12.21 (16.99) |
| Au4 | −33.58 (−29.64) | 19.92 (16.92) | 2.57 (1.37) | 6.81 (5.48) |
| Au5 | −21.11 (−18.48) | 13.86 (13.59) | 13.43 (13.72) | 13.42 (14.36) |
| Au6 | −16.55 (−12.81) | 11.61 (12.23) | 8.14 (5.48) | — |
| Au7 | −16.15 (−13.16) | 9.14 (8.79) | 11.68 (12.22) | 13.92 (15.08) |
| Au8 | −19.66 (−15.58) | 14.35 (11.60) | 6.44 (4.60) | 3.92 (2.99) |
| Au9 | −18.54 (−14.94) | 12.01 (10.88) | 8.90 (5.53) | 12.68 (19.55) |
| Au10 | −16.83 (−14.32) | 13.94 (13.18) | 6.97 (4.28) | 4.49 (4.74) |
The energy barriers of the 5-exo cyclization step are from 9.00 to 14.50 kcal mol−1 for Au5–10 clusters in the gas phase. Those for Au3 and Au4 clusters are 19.00 kcal mol−1. Analysis of the energy barriers of the 5-exo cyclization step and the adsorption energies of the substrate showed that large adsorption energies result in high energy barriers for the 5-exo cyclization step. This point should be attributed to the decreasing interaction between the Cβ atom of substrate fragment and gold clusters in during the 5-exo cyclization step. Hence, the appropriate interactions between the substrate and gold clusters are essential for the 5-exo cyclization step. These findings are also supported by the EDA results (Tables S1–S8†). The ring-opening of the furan step on the Au4–10 clusters is exothermic with moderate energy barriers (Table 2).
The energy barriers of the ring-closing of the dienone carbene–gold step show an interesting “odd–even” behavior respective to the number of gold atoms. Indeed, the energy barriers on odd-numbered gold clusters (Au5, Au7, Au9) are commonly higher than that on gold clusters with even-numbered atoms (Au6, Au8, Au10; Table 2). The different energy barriers on Au3–10 clusters are ascribed to that of the gold clusters with odd numbers of electrons that can strongly bind with the carbene fragment in IM3. Table 2 shows that the rate-determining step is the 5-exo cyclization step on an even-numbered gold clusters, while the rate-limiting step is the ring-closing of dienone carbene–gold intermediate step (IM3 → IM4) with odd-numbered gold clusters as catalysts. This is attributed to the strong interactions between the gold atom and the carbene fragment for the odd-numbered gold clusters as catalysts. The length of the Au–Cα bond in Fig. S3–S9† could not determine this interaction difference. To confirm these findings, we performed EDA on the ring-closing of the dienone carbene–gold intermediate step (IM3 → IM4). As shown in Tables S2–S9,† the interaction energies between the gold clusters and the carbene fragment in IM3 of the odd-numbered gold clusters are larger than those of even-numbered gold clusters. This results in a stronger Au–Cα bond in the odd-numbered gold clusters. The ring-closing of the dienone carbene–gold intermediate step involves the nonequivalent hybridization sp2 orbital of the O atom interaction with the π orbital of the AuCα bond to form a Cα–O bond, which weakens the Au–Cα bond. Therefore, the strong Au–Cα bond of the odd-numbered gold clusters results in large energy barriers.
As shown in Fig. S3–S9,† all of the energy profiles suggest that the planar gold clusters have good catalytic activity toward the ω-alkynylfuran cycloisomerization to the phenol derivative. However, the free energy profiles at 298.15 K and 1 atom indicate that the ω-alkynylfuran cycloisomerization to phenol derivative catalyzed by Au5–10 clusters must overcome energy barriers of about 5.00–18.56 kcal mol−1. Hence, Au3 and Au4 clusters show better catalytic activity toward the ω-alkynylfuran cycloisomerization to phenol derivative than that of Au5–10 clusters at experimental conditions. This is in line with experimental results.18
Comparison with gold(I) complexes
To distinguish the difference between the gold clusters and gold complexes-catalyzed ω-alkynylfuran cycloisomerization, the possible reaction pathways of the ω-alkynylfuran cycloisomerization catalyzed by the [AuP(CH3)3]+ complex was investigated. The reactions were taken from ref. 4 and 10. The P(CH3)3 was used to model the PPh3 to reduce the computational times. The optimized structures, related parameters, potential energy profiles, and relative Gibbs free energies were shown in Fig. S11 and S12.† It was should be noted that the 5-exo FCT pathway toward the phenol derivative P1 and the pathway toward the pyran derivative P3 could be not obtained with our efforts. Simultaneously, the VDA pathway was not considered due to the high barriers of the formation of the gold–vinylidene carbenoid intermediate. Only the DDA pathway toward the phenol derivative P1 and the 6-endo nucleophilic pathway toward the β-alkenylated furan (P2) were shown in Fig. S11 and S12.†As shown in Fig. S11,† the coordination of the ω-alkynylfuran with the [AuP(CH3)3]+ complex is exothermic by about 41.05 (20.48) kcal mol−1 in the gas phase (acetonitrile). The orbital interaction between the alkynyl group and the gold(I) complex are mainly the donation of the electron from the π orbital of the alkynyl group to the unoccupied d orbitals of the gold atom and the back-donation of the electron from the occupied d orbitals of the gold atom to the π* orbital of the alkynyl group. The intramolecular addition of the CC triple bond to the furan ring in IM1-Au(I) slightly endothermic due to the deformation of the aromaticity of the furan ring. The energy barrier is 24.01 (26.47) kcal mol−1 in the gas phase (acetonitrile). Obviously, this step is unfeasible kinetically. Next, the bridged O atom would migrate to the Cα atom in IM2-Au(I) along with the breakage of the C1–O bond to form a gold–benzene oxide complex IM3-Au(I). Due to the twist of the six-membered ring, this step also need overran an energy barrier of 20.91 (26.08) kcal mol−1 in the gas phase (acetonitrile). Finally, the decomposition of IM3-Au(I) is endothermic by about 16 kcal mol−1 in acetonitrile. After consideration of the entropy effect, this step is only endothermicity of 5.27 kcal mol−1 in acetonitrile. The whole reaction pathway need overcome a free energy barrier of 29.81 kcal mol−1 in acetonitrile at 298.15 K.
The pathway of the formation of the β-alkenylated furan (P2) is initialized from the nucleophilic addition of the Cβ atom to the C1 site of the furan ring to produce a spiro-intermediate IM4-Au(I) (the 5-exo cyclization step). This step is slightly exothermic with an energy barrier of 3.54 (8.46) kcal mol−1 in the gas phase (acetonitrile). It should be noted that the pathway toward the phenol derivative P1 from IM4-Au(I) could not be obtained. The relaxed scan of the potential energy profiles at the TPSSh/def2-TZVP levels could not obtain the pathway. Next, the migration of the Cβ atom from the C1 atom to the C2 atom produce IM5-Au(I) with an energy barrier of 10.56 (13.09) kcal mol−1 in the gas phase (acetonitrile). This step is endothermic by about 10 kcal mol−1 because of the repulsion between the C2 and Cβ atoms in IM5-Au(I), which is also certified by the long C2–Cβ bond in IM5-Au(I). Then, the hydrogen atom on the C2 atom transfers to the Cβ atom via a four-membered ring transition state TS5/6-Au(I). This step is the rate-limiting step of this pathway with an energy barrier of 14.45 (15.40) kcal mol−1 in the gas phase (acetonitrile). Finally, the β-alkenylated furan (P2) is detached from IM6-Au(I) with an endothermicity of 10 kcal mol−1 in acetonitrile.
Obviously, the feasible pathway of the ω-alkynylfuran cycloisomerization catalyzed by the [AuP(CH3)3]+ complex is the formation of the β-alkenylated furan (P2), which is not in line with the production distribution of the experiments.2–12 The main product of the ω-alkynylfuran cycloisomerization catalyzed by the [AuP(CH3)3]+ complex should be the phenol derivative P1. To solve this problem, we further consider the reduction of the [AuP(CH3)3]+ complex to AuP(CH3)3 by the terminal alkyne. As shown in Fig. 7, the transfer of the terminal hydrogen of the alkyne group to TfO− (CF3SO3−) was studied. Obviously, the formation of the IM-TfO intermediate should be exothermic due to the strong electrostatic interaction and hydrogen bond, while the change of the free energy in the acetonitrile solvent due to the decrease of the entropy. The hydrogen transfer step must overwhelm an energy barrier of about 10 kcal mol−1 to produce IMx and TfOH. The reduction of the [AuP(CH3)3]+ complex to AuP(CH3)3 by the terminal alkyne need overrun 16 kcal mol−1 of free energy barrier in acetonitrile. Hence, the [AuP(CH3)3]+ complex could be reduced to AuP(CH3)3 in situ by the terminal alkyne. Therefore, all of the possible pathways of the ω-alkynylfuran cycloisomerization catalyzed by AuP(CH3)3 were studied. The optimized structures, related parameters, potential energy profiles, and relative Gibbs free energies were shown in Fig. S13–S15.† It is similar with that of the Au3 cluster; the pathway for the formation of the β-alkenylated furan (P2) is not obtained. As shown in Fig. S13,† the energy barrier of the rate-liming step of the 5-exo FCT pathway catalyzed by AuP(CH3)3 is 14.90 (15.23) kcal mol−1. Simultaneously, the DDA pathway need to overcome an energy barrier of 19.44 (20.82) kcal mol−1. The pathway of the formation of the pyran derivative must surmount an energy barrier of 25.96 (27.61) kcal mol−1. Hence, the most feasible pathway of the ω-alkynylfuran cycloisomerization catalyzed by AuP(CH3)3 is the 5-exo FCT pathway with the formation of the phenol derivative. After incorporation of the reduction of [AuP(CH3)3]+ complex to AuP(CH3)3, the free energy barrier of the 5-exo FCT pathway is about 16 kcal mol−1 in acetonitrile.
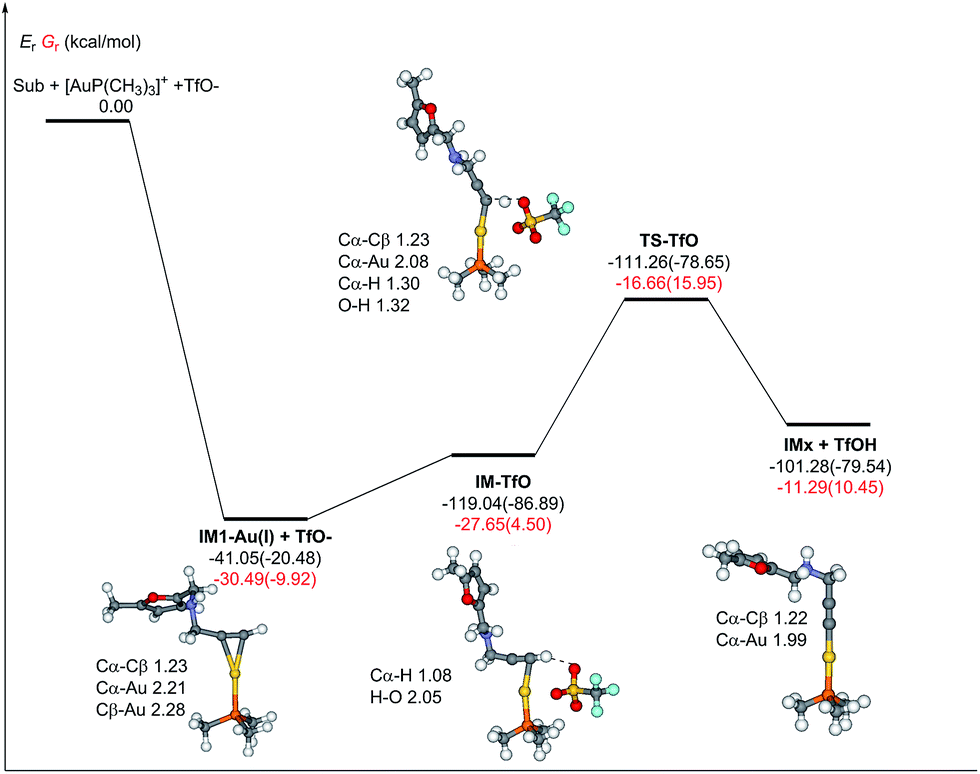 | ||
| Fig. 7 The optimized structures, related parameters, potential energy profile, and relative Gibbs free energies (colored by red, 298.15 K and 1 atm) of the transfer of the terminal hydrogen of the alkyne group to TfO in the gas phase and acetonitrile solvent (parentheses). The bond length is in Å. | ||
Comparison of the potential energy profiles and the free energy profiles in Fig. S11–S15,† it is found that the 5-exo FCT pathway catalyzed by AuP(CH3)3 is the most feasible pathway thermodynamically and kinetically. Then, the pathway of the formation of the β-alkenylated furan (P2) by [AuP(CH3)3]+ is feasible kinetically, however, the whole pathway must overcome about 17.08 kcal mol−1 of free energy barrier at 298.15 K. Therefore, we can conclude that the active catalyst for the ω-alkynylfuran cycloisomerization is the reduction state of the [AuP(CH3)3]+ complex, i.e. AuP(CH3)3. The byproduct (the β-alkenylated furan) is produced from the cyclizaiton of the ω-alkynylfuran catalyzed by the [AuP(CH3)3]+ complex. The selectivity is mainly controlled by the steric effect of the ligand and the polarity of the solvent. The product distribution is in line with the experiments.10 The catalytic activity of the gold(0) complex is comparable with that of the planar gold clusters.
Conclusions
In summary, the full mechanism of the ω-alkynylfuran cycloisomerization catalyzed by a Au3 cluster was investigated at the TPSSh/def2-TZVP levels. Four pathways were identified and analyzed. The 5-exo Friedel–Crafts-type mechanism is the most favorable pathway to produce a phenol derivative with an energy barrier of 19.06 (16.99) kcal mol−1 in the gas phase (acetonitrile). The 6-endo FCT, DDA, and VDA pathways are unfavorable due to the high-energy barriers of the hydrogen migration steps. The strong interaction between gold and vinyl in the Friedel–Crafts adduct stabilizes the Friedel–Crafts addition intermediates. The 5-exo FCT pathways on the planar gold clusters (Au3–10) were studied to clarify the effect size. The appropriate interactions between the substrate and gold clusters are essential for the 5-exo cyclization step. The energy barriers of the ring-closing of the dienone carbene–gold intermediate step show an interesting “odd–even” behavior respective to the number of gold atoms. The Au3 and Au4 clusters show better catalytic activity toward the ω-alkynylfuran cycloisomerization to phenol derivative than the Au5–10 clusters under experimental conditions—this was again confirmed with experimental data. The trade-off studies show that the active catalyst of the ω-alkynylfuran cycloisomerization catalyzed by the gold(I) complexes should be the gold(0) complexes, which is forming in situ reduction by the ω-alkynylfuran. The catalytic activity of the gold(0) complex is comparable with that of the planar gold clusters.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21573030), the Chongqing Science & Technology Commission, China (Grant No. CSTC2013JCYJA50028) and the Scientific Research Foundation of Chongqing University of Arts and Sciences (R2013CJ03). The calculations were performed at the National Supercomputing Center in Shenzhen (Shenzhen Cloud Computing Center).Notes and references
- A. S. K. Hashmi, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553–11554 CrossRef CAS.
- A. S. K. Hashmi, T. M. Frost and J. W. Bats, Org. Lett., 2001, 3, 3769–3771 CrossRef CAS PubMed.
- A. S. K. Hashmi, J. P. Weyrauch, M. Rudolph and E. Kurpejovic, Angew. Chem., Int. Ed., 2004, 43, 6545–6547 CrossRef CAS PubMed.
- A. S. K. Hashmi, M. C. Blanco, E. Kurpejovic, W. Frey and J. W. Bats, Adv. Synth. Catal., 2006, 348, 709–713 CrossRef CAS.
- A. S. K. Hashmi, R. Salathe and W. Frey, Chem.–Eur. J., 2006, 12, 6991–6996 CrossRef CAS PubMed.
- A. S. K. Hashmi, J. P. Weyrauch, E. Kurpejovic, T. M. Frost, B. Frey, W. Miehlich and J. W. Bats, Chem.–Eur. J., 2006, 12, 5806–5814 CrossRef CAS PubMed.
- A. S. K. Hashmi, E. Kurpejovic, M. Wolfle, W. Frey and J. W. Bats, Adv. Synth. Catal., 2007, 349, 1743–1750 CrossRef CAS.
- A. S. K. Hashmi, M. Rudolph, J. W. Bats, W. Frey, F. Rominger and T. Oeser, Chem.–Eur. J., 2008, 14, 6672–6678 CrossRef CAS PubMed.
- Y. Chen, W. Yan, N. G. Akhmedov and X. Shi, Org. Lett., 2010, 12, 344–347 CrossRef CAS PubMed.
- A. S. K. Hashmi, J. Hofmann, S. Shi, A. Schutz, M. Rudolph, C. Lothschutz, M. Wieteck, M. Buhrle, M. Wolfle and F. Rominger, Chem.–Eur. J., 2013, 19, 382–389 CrossRef CAS PubMed.
- M. C. B. Jaimes, C. R. N. Bohling, J. M. Serrano-Becerra and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 7963–7966 CrossRef PubMed.
- M. C. B. Jaimes, F. Rominger, M. M. Pereira, R. M. B. Carrilho, S. A. C. Carabineiro and A. S. K. Hashmi, Chem. Commun., 2014, 50, 4937–4940 RSC.
- B. Martin-Matute, D. J. Cardenas and A. M. Echavarren, Angew. Chem., Int. Ed., 2001, 40, 4754–4757 CrossRef CAS PubMed.
- B. Martin-Matute, C. Nevado, D. J. Cardenas and A. M. Echavarren, J. Am. Chem. Soc., 2003, 125, 5757–5766 CrossRef CAS PubMed.
- S. Carrettin, M. C. Blanco, A. Corma and A. S. K. Hashmi, Adv. Synth. Catal., 2006, 348, 1283–1288 CrossRef CAS.
- A. S. K. Hashmi, M. Rudolph, H.-U. Siehl, M. Tanaka, J. W. Bats and W. Frey, Chem.–Eur. J., 2008, 14, 3703–3708 CrossRef CAS PubMed.
- A. S. K. Hashmi, M. Rudolph, J. P. Weyrauch, M. Wolfle, W. Frey and J. W. Bats, Angew. Chem., Int. Ed., 2005, 44, 2798–2801 CrossRef CAS PubMed.
- J. Oliver-Meseguer, A. Leyva-Perez and A. Corma, ChemCatChem, 2013, 5, 3509–3515 CrossRef CAS.
- J. Oliver-Meseguer, J. R. Cabrero-Antonino, I. Dominguez, A. Leyva-Perez and A. Corma, Science, 2012, 338, 1452–1455 CrossRef CAS PubMed.
- A. S. K. Hashmi, Science, 2012, 338, 1434 CrossRef CAS PubMed.
- J. Oliver-Meseguer, A. Leyva-Perez, S. I. Al-Resayes and A. Corma, Chem. Commun., 2013, 49, 7782–7784 RSC.
- D. Tang, Z. Chen, Y. Tang, J. Zhang, Z. Xu and J. Zhang, J. Phys. Chem. C, 2014, 118, 18510–18520 CrossRef CAS.
- D. Tang, Z. Chen, J. Zhang, Y. Tang and Z. Xu, Organometallics, 2014, 33, 6633–6642 CrossRef CAS.
- J. M. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- K. P. Jensen, Inorg. Chem., 2008, 47, 10357–10365 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Phys., 2004, 120, 9918–9924 CrossRef CAS PubMed.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61–69 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- Y.-K. Shi, Z. H. Li and K.-N. Fan, J. Phys. Chem. A, 2010, 114, 10297–10308 CrossRef CAS PubMed.
- L. Li, Y. Gao, H. Li, Y. Zhao, Y. Pei, Z. Chen and X. C. Zeng, J. Am. Chem. Soc., 2013, 135, 19336–19346 CrossRef CAS PubMed.
- C. Liu, Y. Tan, S. Lin, H. Li, X. Wu, L. Li, Y. Pei and X. C. Zeng, J. Am. Chem. Soc., 2013, 135, 2583–2595 CrossRef CAS PubMed.
- S. Lin and Y. Pei, J. Phys. Chem. C, 2014, 118, 20346–20356 CrossRef CAS.
- M. Boronat, T. Loópez-Ausens and A. Corma, J. Phys. Chem. C, 2014, 118, 9018–9029 CrossRef CAS.
- R.-J. Lin, H.-L. Chen, S.-P. Ju, F. Y. Li and H.-T. Chen, J. Phys. Chem. C, 2012, 116, 336–342 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- N. E. Schultz, Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 11127–11143 CrossRef CAS PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef PubMed.
- T. M. Soini, A. Genest, A. Nikodem and N. Rösch, J. Chem. Theory Comput., 2014, 10, 4408–4416 CrossRef CAS PubMed.
- P. Janthon, S. Luo, S. M. Kozlov, F. Viñes, J. Limtrakul, D. G. Truhlar and F. Illas, J. Chem. Theory Comput., 2014, 10, 3832–3839 CrossRef CAS PubMed.
- B. Chan and W.-L. Yim, J. Chem. Theory Comput., 2013, 9, 1964–1970 CrossRef CAS PubMed.
- G. A. Bishea and M. D. Morse, J. Chem. Phys., 1991, 95, 5646–5659 CrossRef CAS.
- A. M. James, P. Kowalczyk, B. Simard, J. C. Pinegar and M. D. Morse, J. Mol. Spectrosc., 1994, 168, 248–257 CrossRef CAS.
- K. Hilpert and K. A. Gingerich, Ber. Bunsen-Ges. Phys. Chem., 1980, 84, 739–745 CrossRef CAS.
- B. Assadollahzadeha and P. Schwerdtfeger, J. Chem. Phys., 2009, 131, 064306 CrossRef PubMed.
- S. A. Serapian, M. J. Bearpark and F. Bresme, Nanoscale, 2013, 5, 6445–6457 RSC.
- M. P. Johansson, I. Warnke, A. Le and F. Furche, J. Phys. Chem. C, 2014, 118, 29370–29377 CrossRef CAS.
- G. A. Bishea and M. D. Morse, J. Chem. Phys., 1991, 95, 8779–8792 CrossRef CAS.
- A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organometallics, 2012, 31, 644–661 CrossRef CAS.
- A. S. K. Hashmi, M. Wieteck, I. Braun, P. Nosel, L. Jongbloed, M. Rudolph and F. Rominger, Adv. Synth. Catal., 2012, 354, 555–562 CrossRef CAS.
- A. S. K. Hashmi, M. Wieteck, I. Braun, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 10633–10637 CrossRef CAS PubMed.
- M. M. Hansmann, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 2593–2598 CrossRef CAS PubMed.
- M. H. Vilhelmsen and A. S. K. Hashmi, Chem.–Eur. J., 2014, 20, 1901–1908 CrossRef CAS PubMed.
- J. Bucher, T. Wurm, K. S. Nalivela, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2014, 53, 3854–3858 CrossRef CAS PubMed.
- M. M. Hansmann, F. Rominger and A. S. K. Hashmi, Chem. Sci., 2013, 04, 1552–1559 RSC.
- B. Hammer, Surf. Sci., 2000, 459, 323–348 CrossRef CAS.
- C. Dupont, Y. Jugnet and D. Loffreda, J. Am. Chem. Soc., 2006, 128, 9129–9136 CrossRef CAS PubMed.
- Z. Wei, D. Li, X. Pang, C. Lv and G. Wang, ChemCatChem, 2012, 4, 100–111 CrossRef CAS.
- Y. C. Choi, W. Y. Kim, H. M. Lee and K. S. Kim, J. Chem. Theory Comput., 2009, 5, 1216–1223 CrossRef CAS PubMed.
- R. M. Olson, S. Varganov, M. S. Gordon, H. Metiu, S. Chretien, P. Piecuch, K. Kowalski, S. A. Kucharski and M. Musial, J. Am. Chem. Soc., 2005, 127, 1049–1052 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S15, Tables S1–S9, details of the energy decomposition analysis, and the Cartesian coordinates, single point electronic energies (in the gas phase and acetonitrile), zero-point, and free energy correction for all of the stationary points and imaginary frequencies of transition structures. See DOI: 10.1039/c5ra26156b |
| This journal is © The Royal Society of Chemistry 2016 |