DOI:
10.1039/C5RA26079E
(Paper)
RSC Adv., 2016,
6, 9958-9966
Improving KNbO3 photocatalytic activity under visible light†
Received
7th December 2015
, Accepted 30th December 2015
First published on 6th January 2016
Abstract
An increasing number of photocatalytic applications of KNbO3 in different fields motivated us to find an efficient strategy to reduce its band gap so that it can utilize the solar spectrum. Using density functional theory (DFT) with a hybrid functional proposed by Heyd, Scuseria, and Ernzerhof, the experimental band gap (3.24 eV) of KNbO3 was successfully reproduced (3.23 eV). In the present study, we systematically investigated the effect of doping with N and W on the geometry and electronic structure of KNbO3. Because of the closeness of the ionic radius small changes in the parent crystal structure occurs. However, the electronic structure showed major changes in both the cases. N introduces impurity states adjacent to the top of the valence band and the bottom of the conduction band (CB), thus reducing the band gap significantly. Doping with W results in an n-type semiconductor, and introduces occupied states adjacent to the CB. Although both the dopant elements can improve the visible light absorption, it may accelerate electron–hole recombination. Therefore, individually they may not be able to improve the photocatalytic activity of KNbO3. Interestingly, a highly favourable band structure was produced with a reduced band gap when both N and W are simultaneously doped into the crystal structure of KNbO3. The calculated formation energy indicates that the doping of N becomes more feasible in the presence of W. This may be due to the formation of a charge compensated system, which also reduces the vacancy defect formation. More importantly, the band edge shifting in the presence of both N and W occurs in such a controlled fashion that KNbO3 still remains suitable for overall water splitting. Therefore, one can justify the choice of the (N, W) pair for improving the visible light driven photoactivity of KNbO3.
1. Introduction
Development of alternate sources of energy has become one of the prime research areas due to limited content of fossil fuels and their impact on the environment. Extensive efforts have been dedicated over the previous decades to find efficient strategy for conversion of solar energy to usable energy. Among them, the generation of hydrogen through photocatalytic splitting of water under sunlight is considered as a promising approach.1–6 The major challenge in this area is to find a suitable material for this purpose. An ideal catalyst should have absorption characteristics in the visible range of the solar spectrum, favorable crystal morphology for high photoconversion efficiency, and proper band edge alignment for spontaneous generation of hydrogen and oxygen during water splitting. However, it is difficult to find a single material satisfying all these properties. Among different catalysts developed to date, metal oxide based semiconductor materials hold a large area. Metal oxide materials of the perovskite type have generated immense interest due to several unique features, such as long term durability, wide availability, non-toxicity and an easy to tune electronic structure depending on the requirement.6–13 Recently, KNbO3 has been studied by several groups due to its potential applications in various photocatalytic processes, including hydrogen generation though water splitting, degradation of organic pollutants, and CO2 conversion. KNbO3 exists mainly in three different crystalline phases, viz., cubic, orthorhombic, and tetragonal.14,15 The photocatalytic property has been shown to strongly depend on the crystal structure. In a recent experimental study by Zhang et al., it was shown that the cubic form shows the highest photocatalytic activity due to having the highest symmetry in the bulk structure and an associated favourable electronic structure.14 The major drawback for all the forms is their poor photoactivity under visible light exposer due to a large band gap (3.24, 3.15, and 3.08 eV, respectively).14 To date, various strategies have been adopted for improving the photocatalytic activity of KNbO3. As for example, Ding et al. synthesized KNbO3 nanowire using a hydrothermal method and obtained an enhanced photocatalytic activity for water splitting.16 They indicated that increased surface area, good crystallinity, and fineness of the KNbO3 nanowire are responsible for improved quantum efficiency of water splitting activity. Recently, Zhang et al. investigated the structure–photoactivity relationship in KNbO3 nanowires using scanning transmission electron spectroscopy combined with first principles calculations.17 They concluded that the difference in the reducing behaviour due to variation in the conduction band minimum (CBM) potential and the extent of electron–hole separation results in different photoactivity for the various KNbO3 polymorphs. In a recent study, different KNbO3 nanostructures have been synthesized, including nanowires, nanotowers, nanocubes and nanorods by controlling reactant concentration, temperature, and reaction time.18 Among the polymorphs, nanocubes showed the highest photoactivity due to high crystallinity and active surface facets. One dimensional KNbO3 nanowire with Au nanoparticle anchoring onto it has been found to show better photocatalytic response than the commercial variety.19 The crucial role of surface plasmon resonance and interband transition on gold nanoparticles is responsible for the improved photocatalytic activity. Yan et al. utilized similar phenomena by synthesizing a KNbO3 microcube deposited with gold nanoparticles, which shows enhanced photocatalytic activity under visible light.20 An enhancement of the optical property of KNbO3 was observed by Wang et al. due to doping with Na, which reduces the band gap to 3.09 eV.21 The perturbation of the energy levels associated with lattice distortion and mass effect due to introduction of Na at the K lattice site was found to be responsible for the change in band gap. Interestingly, the optical property of Na–K mixed niobates has been found to strongly depend on the different polymerization agent involved during synthesis.22 Several attempts by doping with various lanthanide and actinide elements have been made to change the optical property of KNbO3.23,24 Guo et al. observed enhanced KNbO3 photocatalytic activity of under visible light towards hydrogen evolution by using Er3+:Y3Al5O12, which is an upconverting luminescence agent.25 In a recent study, Wang et al. demonstrated hydrogen production under visible light using a heterojunction consisting of self (Nb4+)-doped KNbO3 and Nb4N5.26 In this case, the heterojunction facilitates separation of photogenerated electron and holes and the Nb4N5 unit acts as a cocatalyst, leading to improved photocatalytic response. A significant improvement of visible light activity of KNbO3 has been observed in the study of Wang et al. by N doping.27 It is well known that N doping into oxide based semiconducting materials has several disadvantages, leading to faster recombination rates.28–32 To overcome this, a number of strategies have been developed based on codoping with anionic or cationic dopant elements.33–37 In this study, we investigated the effect of introducing a cationic dopant on the electronic structure of N-doped KNbO3. To select a suitable codopant the criteria that were considered are as follows: (i) the element should form a charge compensated system, so that vacancy defect formation can be at a minimum; (ii) the ionic size of the dopant element should be such that it can easily be fitted into the host lattice site; and (iii) the dopant element should not lower the CBM by a significant extent to maintain KNbO3 active for hydrogen evolution. Based on these criterion, we choose W as a potential codopant for the N-doped KNbO3. It is not expected to cause a CBM downward shift by a large extent in the presence of W at the Nb lattice site, because the W 5d-orbital is energetically higher than the Nb 4d orbital.38 To obtain a detailed description of the band structure of KNbO3 in the presence of both N and W, we performed calculations using the Heyd, Scuseria, and Ernzerhof (HSE) hybrid functional,39 which has been found to successfully overcome the limitations of standard density functional theory (DFT) for wide range of semiconductor materials.40,41 The present study also addressed the effect of individual dopant elements on the KNbO3 electronic structure.
We present a complete description of the geometry and electronic structure of KNbO3 in the absence and presence of dopant element, and the band edge alignment with respect to water redox levels. Details of the calculation methodology used to obtain all of this information are presented as well. Finally, some concluding remarks are given based on the present investigation.
2. Computational details
All the description related to geometry and electronic structure presented herein were based on calculations using the projector augmented wave (PAW)42 based code, Vienna ab initio simulation package (VASP).43 The doped systems were modelled using a 2 × 2 × 2 supercell of cubic KNbO3 crystal structure (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, space group no. 221). Larger supercells were also encountered to achieve lower concentrations. All the structures were fully relaxed (both cell parameter and ionic position) to obtain the equilibrium geometry. A Monkhorst and Pack scheme44 was employed to integrate a Brillouin zone with sufficiently large number of k-point mesh (8 × 8 × 8). The self-consistent iteration was continued until the energy convergence reached 10−6 eV. Geometry optimization was carried out with an exchange correlation potential of Perdew–Burke–Ernzerhof (PBE) under a generalized gradient approximation (GGA).45 To define the interactions between the ionic core and valence electrons, the PAW potential was considered. The sets of valence electrons involved in the pseudopotential were K (3p64s1), Nb (4p65s24d3), W (6s25d4), O (2s22p4), and N (2s22p3). The valence electron orbital was expanded in the plane wave basis set up to a sufficiently large Ecut value (500 eV). The calculation for the electronic structure was carried out using the HSE hybrid functional.39 In this approach, the exchange functional was decomposed into short range (SR) and long range (LR) parts. The long range part is completely described by the PBE functional, whereas the short range part by both PBE and exact Hartree–Fock (HF) exchange. The mathematical form for the exchange correlation functional is given as follows:
m, space group no. 221). Larger supercells were also encountered to achieve lower concentrations. All the structures were fully relaxed (both cell parameter and ionic position) to obtain the equilibrium geometry. A Monkhorst and Pack scheme44 was employed to integrate a Brillouin zone with sufficiently large number of k-point mesh (8 × 8 × 8). The self-consistent iteration was continued until the energy convergence reached 10−6 eV. Geometry optimization was carried out with an exchange correlation potential of Perdew–Burke–Ernzerhof (PBE) under a generalized gradient approximation (GGA).45 To define the interactions between the ionic core and valence electrons, the PAW potential was considered. The sets of valence electrons involved in the pseudopotential were K (3p64s1), Nb (4p65s24d3), W (6s25d4), O (2s22p4), and N (2s22p3). The valence electron orbital was expanded in the plane wave basis set up to a sufficiently large Ecut value (500 eV). The calculation for the electronic structure was carried out using the HSE hybrid functional.39 In this approach, the exchange functional was decomposed into short range (SR) and long range (LR) parts. The long range part is completely described by the PBE functional, whereas the short range part by both PBE and exact Hartree–Fock (HF) exchange. The mathematical form for the exchange correlation functional is given as follows:| | |
EHSEXC = aESRX(μ) + (1 − a)EPBE, SRX(μ) + EPBE, LRX(μ) + EPBEC
| (1) |
where ‘μ’ is the screening parameter, dictating the length of the SR and LR parts. In the present study, the value of screening parameter was kept fixed at 0.2 Å−1. The extent of mixing was defined by the mixing coefficient ‘a’. Herein, a set of test calculations have been carried out on undoped KNbO3 using various values of the mixing parameter to get a better match of the calculated band gap with the experimental value (3.24 eV).14 The optimized parameter was employed for the electronic structure calculations of all the doped and codoped systems. The band structure was shown along the high symmetry K-path. For density of state (DOS) calculations, the tetrahedron method with a Blöchl correction was employed.46
3. Results and discussion
3.1. Geometry
KNbO3 exists in different crystal structures such as cubic, orthorhombic, and tetragonal. Recent studies have shown that the photocatalytic activity of the cubic form is much higher than that of the other two due to high symmetry in the crystal structure and a favourable electronic structure.14 Therefore, in this study, all the calculations were carried out using cubic KNbO3. As shown in Fig. 1, the cationic atom K and Nb are distributed on a simple cubic lattice and the anionic atom O occupies the face centers nearest to Nb. Thus, Nb locates itself at the centre of each oxygen octahedral, whereas K remains at the twelve fold coordinated sites. The lattice parameter for the 2 × 2 × 2 cell was found to be 8.125 Å at the GGA_PBE calculation level. The Nb–O bond length was 2.03 Å and Nb–O–Nb bond angle exactly equalled 180°. Doping with N results in a small change in the parent crystal structure due to the closeness of its ionic size with that of oxygen (RO2−= 1.35 Å, RN3− = 1.46 Å).47 However, the introduction of W at the Nb lattice site increased the cell length by 0.29 Å along the c-direction. The W–O bond length was found to be relatively shorter (1.81 Å) than the Nb–O length in the ideal crystal structure. This increases the adjacent Nb–O distance by 0.46 Å. The bond angles (W–O–Nb and Nb–O–Nb) perpendicularly cutting the c-axis were bent to around 170°, although the angle parallel to the c-axis remains as it is (180°). The cell volume increases by 9 Å3. In the case of codoping, both N and W are simultaneously introduced into the crystal structure of KNbO3. To consider different configurations for the codoped systems we chose different lattice sites for the dopant pair. In this case, one can generate two different configurations, which are structurally inequivalent to each other. The dopant elements may occupy a nearest lattice site (Nb1, O1) or one far away from each other (Nb1, O2) in Fig. 1, resulting in the formation of different configurations, labelled ‘Str. 1’ and ‘Str. 2’, respectively. In this case, ‘Str. 1’ is energetically more stable by 0.59 eV than the ‘Str. 2’. Depending upon the synthetic strategy, the configuration of the codoped system may be different. For example, a synthetic route like solution method preferably produces an energetically more stable structure,48 whereas techniques such as magnetron sputtering or supersonic cluster beam deposition, generate structures that may not be the lowest energy configurations.49–51 Therefore, in the present study both type of configurations were considered. In the optimized geometries, the calculated value of the distance between the two dopant elements was found to be 1.75 and 4.45 Å in the two structures. The cell volume for both configurations was found to be almost equal. In case of ‘Str. 1’, the cell length increased by 0.5 Å in the c-direction, whereas the same was observed along b-direction for ‘Str. 2’. The M–O–M′ (M = Nb, M′ = Nb, or W) bond angle perpendicular to the c-axis is bent to 165° for ‘Str. 1’, whereas in ‘Str. 2’ it occurred for the same bond angle parallel to the c-axis. In the following section, it can be observed how this variation of dopant–dopant separation influences the electronic structure of (N, W)-codoped KNbO3. But before that the feasibility of formation of the codoped systems by considering their stability with respect to the individual doping process will be discussed.
 |
| | Fig. 1 A 2 × 2 × 2 supercell of KNbO3 cubic crystal structure. The number indicates the position of the dopant element. | |
3.2. Defect formation energy
To find out the feasibility of doping and codoping into the crystal structure of KNbO3 the calculation the defect formation energy was carried out. In the present study, we also investigated the variation of the defect formation energy as a function of the host chemical potential to explore the most suitable environment for synthesis of the doped or codoped materials. The expression for the defect formation energy can be written as follows:52,53| | |
ΔHf = Edoped − EKNbO3 + nOμO + nNbμNb − nNμN − nWμW
| (2) |
where EKNbO3 and Edoped stand for the energy of the undoped and doped or codoped KNbO3, respectively, nA represents the number of elements substituted or introduced during the process of doping, and μA indicates the chemical potential of the host and guest elements.
At the condition of equilibrium between the reservoir of K, Nb, O and KNbO3, the relation is as follows:
| | |
μK + μNb + 3μO = μKNbO3(bulk).
| (3) |
The chemical potentials of the constituent elements satisfy by the relations, μK ≤ μK(bulk), μNb ≤ μNb(bulk), and μO ≤ μO(gas).
The heat of formation (Δ) for bulk KNbO3 can be calculated using the relation:
| | |
Δ = μKNbO3(bulk) − μK(bulk) − μNb(bulk) − 3μO(gas)
| (4) |
Because KNbO3 is a stable material, Δ is a negative quantity. Therefore, one can define the ranges of μK, μNb, and μO as follows:
| | |
μK(bulk) + Δ ≤ μK ≤ μK(bulk)
| (5a) |
| | |
μNb(bulk) + Δ ≤ μNb ≤ μNb(bulk)
| (5b) |
| | |
3μO(gas) + Δ ≤ 3μO ≤ 3μO(gas)
| (5c) |
herein,
μK(bulk),
μNb(bulk), and
μW are calculated from the energy of an atom in the respective bulk metallic state. On the other hand,
μO(gas) and
μN have been obtained by calculating the energy of the diatomic gaseous molecule at the center of a 20 Å × 20 Å × 20 Å cubic box (
μO(gas)/N = 1/2
EO2/N2).
The variation of the defect formation energy as a function of chemical potential of the host is shown in Fig. 2. Herein, ΔμA = 0 indicates an extreme A-rich condition. As can be observed from Fig. 2a and b, formation of N-doped and W-doped KNbO3 was more feasible under host poor conditions. This may be due to difficulty in vacancy formation under host rich conditions. Comparing Fig. 2a and b indicates that the doping with N is relatively less favourable than W-doping. For codoping variation of defect formation energy has been plotted as a function of both μO and μNb. Fig. 2c shows the defect formation energy profile for both ‘Str. 1’ (solid lines) and ‘Str. 2’ (dotted lines). The defect formation energy for the codoped system was found to be decreased compared to that of the N-doped system, i.e., in presence of W, the doping of N becomes energetically more feasible. The formation of a charge compensated system may be the driving force for this. As can be observed from Fig. 2c, the formation energy for (N, W)-codoped KNbO3 with ‘Str. 1’ was lower than that of ‘Str. 2’, indicating preferable formation of ‘Str. 1’ under equilibrium conditions. For both cases, the O-rich condition was found to be more favourable than the Nb-rich condition.
 |
| | Fig. 2 The variation of defect formation energy with the chemical potential of O (μO − μO-gas) and Nb (μNb − μNb-bulk) for N-doped KNbO3 (N: 4.17%) (a), W-doped KNbO3 (W: 12.5%) (b) and (N, W)-codoped KNbO3 (N: 4.17%, W: 12.5%) (c). The color lines (c) corresponds to different formation energies for the (N, W)-codoped KNbO3. The codoped system cannot be formed in the lower half of the diagonal axis (large white region) in the case of (c) because the chemical potential of K (μK) exceeded (eqn (5a)). | |
3.3. Electronic structure
In this section, we discuss the role of dopant element is modifying the band structure of KNbO3. For this purpose, several test calculations were performed with undoped KNbO3 by varying the computational parameters under the HSE formalism. The PBE0 hybrid functional was also taken into account. As can be observed from Table 1, the calculated band gap with a 0.2 Å−1 screening parameter and 36% of exact HF mixing exchange showed the best match (3.23 eV) with the experimentally reported band gap of KNbO3 (3.24 eV).14 Therefore, throughout the calculations, we employed this computational parameter for investigating the electronic structure of the doped and codoped systems. Fig. 3 shows the band structure plot for the undoped KNbO3 along the high symmetry K-path. Analysis of PDOS indicated that the valence band (VB) is composed of O 2p state, whereas the conduction band (CB) is contributed by Nb 4d state. This is similar to the characteristics of typical metal oxide based materials. Absence of K related states in the band edge is the consequence of ionic type bonding between the K and NbO6 units. Furthermore, how this band structure is influenced in presence of the dopant element is discussed by describing the electronic structure first in the presence of a single dopant element, followed by the presence of both the dopant elements.
Table 1 Variation of the band gap of undoped KNbO3 with different computational parameters. Experiment data taken from ref. 14
| Mixing HF exchange (%) |
Screen parameter (Å−1) |
Band gap (eV) |
| 25 |
0.2 |
2.62 |
| 30 |
0.2 |
2.89 |
| 35 |
0.2 |
3.17 |
| 36 |
0.2 |
3.23 |
| PBE0 |
3.34 |
| Experimental14 |
3.24 |
 |
| | Fig. 3 Band structure and density of states of undoped KNbO3. The horizontal line in the band structure plot and vertical line in the density of states plot indicates the Fermi level. | |
3.3.1. N-doped KNbO3. Doping with N at the oxygen lattice site results in significant changes in the KNbO3 electronic structure. The band structure plot (Fig. 4a) shows the presence of new states at both band edges. Therefore, the effective band gap was reduced to 1.91 eV, which can lead to significant visible light activity enhancement. It is interesting to observe that the new states appeared adjacent to the band edges, instead of as discrete states in the forbidden region, which occurs for most of N-doped metal oxide materials.9,37 The DOS plot (Fig. 4a′) was no longer symmetric with respect to spin up and spin down parts. The impurity states near the VB were partially occupied. This is due to a one electron deficiency created by N doping that contains one less electron in the valence shell (2s22p3) than that of oxygen (2s22p4), leading to paramagnetism (magnetic moment 1 μB per supercell). To obtain a detailed description of the impurity states we analysed the PDOS. It can be observed from Fig. 4a′ that N 2p and O 2p hybridized states contribute to the VBM of N-doped KNbO3, whereas the states at the CB edge are composed of O 2p, N 2p and Nb 4d states. One of the main factors behind the change is the higher energy of N 2p orbital with respect to the O 2p orbital and electron deficiency. The difference in electronegativity between nitrogen and oxygen may also influence the bonding with Nb. It is well known that doping of N into metal oxide semiconductor improves the visible light activity by a significant extent. However, the reported photoconversion efficiency was found to be far below the expected level.28–32 In this case, the partial occupancy of the states may be harmful for longer lifetime of the photogenerated charge carriers. Another major issue is the spontaneous formation of charge compensating vacancy defects, which can accelerate the electron–hole recombination. However, impurity states at the CB lower the CBM by a significant extent (0.56 eV), which may in turn lower the reducing behaviour (hydrogen evolution reaction during water splitting). Therefore, to improve the photocatalytic activity of N-doped KNbO3, one needs to introduce another dopant element, which can compensate the electron deficiency and form a charge compensated system. For this purpose, we choose W as a codopant of N-doped KNbO3. In the next section, a description of the electronic structure of KNbO3 only in presence of W is presented.
 |
| | Fig. 4 Band structure (a) and density of states (a′) of N-doped KNbO3 (N: 4.17%). The horizontal line in (a) and vertical line in (a′) indicates the Fermi level. | |
3.3.2. W-doped KNbO3. Doping of W at the Nb lattice site results into an n-type semiconductor. The band structure plot (Fig. 5a) shows the presence of Fermi level in the CB region. Calculation of the magnetic moment indicates that the total magnetic moment for W-doped KNbO3 is 0.8 μB per supercell. The excess electron environment is created due to the presence of one more electron in the valence shell W than that of Nb. This leads to occupied impurity states adjacent to the bottom of the CB, and consequently the CBM is lowered by 0.86 eV with respect to that of undoped KNbO3, leading to reduction in band gap to 2.21 eV. Therefore, significant visible light activity enhancement is also expected in this case. However, lowering of CBM by such a large extent and the associated charge compensating defects are the main limiting factors for W-doped KNbO3. Because of unpaired electron, the DOS plot (Fig. 5a′) is unsymmetrical with respect to the spin up and spin down parts. To obtain information about the contributory states at the band edge we analysed the PDOS. As can be observed from Fig. 5a′, the VBM is composed of O 2p states, whereas the partially occupied impurity states near the CB are the hybridized states of the Nb 4d and W 5d states. Therefore, codoping of W into the crystal structure of N-doped KNbO3 is expected to compensate for the electron deficiency. In the next section, the role of both N and W on the electronic structure of KNbO3 is presented.
 |
| | Fig. 5 Band structure (a) and density of states (a′) of W-doped KNbO3 (W: 12.5%). The horizontal line in (a) and vertical line in (a′) indicates the Fermi level. | |
3.3.3. (N, W)-codoped KNbO3. Herein, we have considered two different possible configurations for the (N, W)-codoped KNbO3, which differ in the length of dopant–dopant separation. The probability of forming a particular type of product strongly depends on the choice of synthetic strategy. In the presence of both N and W, the total electrical charge of the undoped system is maintained. Fig. 6 shows that a clean band structure is produced for (N, W)-codoped KNbO3, wherein the Fermi level is located on the top of the VB, similar to the case of an intrinsic semiconductor. A considerable reduction in the band gap was observed in case of ‘Str. 2’ (0.47 eV), whereas for ‘Str. 1’ the reduction was rather small (0.1 eV). Analysis of electronic energy levels indicates that the band gap varied due to the difference in relative shift of the VBM and CBM. For ‘Str. 1’ the VBM was found to be elevated only by a small extent (0.07 eV), whereas the CBM remains almost unchanged with respect to that of undoped KNbO3. However, in case of ‘Str. 2’, both the VBM and CBM were elevated by 0.52, and 0.05 eV, respectively. The important observation is that in both cases the band gap is controlled by the upward shifting of the VBM only, leaving the CBM almost unchanged. Therefore, the present strategy of narrowing the band gap does not sacrifice the reducing behaviour of KNbO3 at the CB. This is very important for narrowing the band gap of a hydrogen evolving photocatalyst. In both the cases, the DOS plot (Fig. 6a′ and b′) was symmetrical with respect to spin up and spin down parts. To investigate the electronic structure for both the configurations in details, we analyze the PDOS. As can be observed from Fig. 6, the nature of VBM and CBM are similar in both cases. The VBM is composed of hybridized N 2p and O 2p states, whereas the CBM is contributed by a mixed state of Nb 4d and W 5d states. Investigating the change in electronic charge distribution in presence of dopant element requires analysis of the charge density plot (Fig. 7). As can be observed from Fig. 7a, the charge distribution is almost symmetrical in the case of undoped KNbO3. In the presence of W (Fig. 7b), the symmetric nature was lost, indicating less covalent character between the W–O bond. Comparing the charge density plot for the ‘Str. 1’ and ‘Str. 2’ (Fig. 7c and d), indicating that in the latter case N is more covalently bonded to Nb than in the first case. This may have important influence on the difference in electronic energy levels of the two different configurations for the codoped systems. Therefore, based on this present investigation, we conclude that improvement of visible light activity of KNbO3 by codoping with N and W can only be achieved by choosing an appropriate synthetic strategy, such as magnetron sputtering or supersonic cluster beam deposition, which is expected to deliver materials of the type ‘Str. 2’ as a major product.49–51
 |
| | Fig. 6 Band structure and density of states of (N, W)-codoped KNbO3 (N: 4.17%, W: 12.5%) using Str. 1 (a and a′) and Str. 2 (b and b′). The horizontal line in the band structure plot and vertical line in the density of states plot indicates the Fermi level. | |
 |
| | Fig. 7 Charge density distribution for undoped KNbO3 (a), W-doped KNbO3 (W: 12.5%) (b), (N, W)-codoped KNbO3 (N: 4.17%, W: 12.5%) with Str. 1 (c) and Str. 2 (d). | |
3.4. Effect of concentration
To investigate the effect of reduced dopant concentration on the electronic structure of (N, W)-codoped KNbO3, calculations with a larger supercell size, 2 × 2 × 3 (dopant concentration for N: 2.78%, W: 8.33%) and 2 × 3 × 3 (dopant concentration for N: 1.85%, W: 5.55%), have been carried out. The dopant concentration stands for the atom percentage. In a KNbO3 unit cell, there exists one Nb lattice site, whereas number of oxygen in the lattice is three, this leads to different concentration for N and W in the same supercell. For the 2 × 2 × 3 case, two different configurations were taken into account by placing the dopant elements either at the (Nb1, O1) or (Nb1, O2) lattice site in Fig. S1,† which are denoted as ‘Str. 1’ and ‘Str. 2’, respectively. Similarly in the case of the 2 × 3 × 3 supercell (Fig. S2†), ‘Str. 1’ (Nb1, O1) and ‘Str. 2’ (Nb1, O2) have been considered. All the structures have been fully optimized, i.e., relaxation of both ionic positions as well as the cell parameter has been considered. Brillouin zone sampling was carried out with a k-point mesh of 6 × 6 × 4 and 6 × 4 × 4 for the 2 × 2 × 3 and 2 × 3 × 3 supercell, respectively. In the optimized geometries of 2 × 2 × 3 case (‘Str. 1’, and ‘Str. 2’), the calculated distance between N and W was 1.75, and 7.33 Å, respectively. In this case, ‘Str. 1’ is energetically more stable than ‘Str. 2’ by 1.32 eV. The DOS plot was presented in Fig. S1,† which shows that the nature of band structure is almost similar to the respective higher concentration cases. For ‘Str. 1’, the calculated band gap is 3.16 eV, which is only 0.02 eV higher than that calculated using ‘Str. 1’ of a 2 × 2 × 2 supercell. However, for ‘Str. 2’, the decrease in band gap was found to be more significant (0.87 eV) than the corresponding higher concentration case. The difference may be due to increased dopant–dopant separation (7.33 Å). In the case of the 2 × 3 × 3 supercell, ‘Str. 1’ was found to be energetically more stable by 0.74 eV than the ‘Str. 2’. The calculated distance between the dopant elements in the 2 × 3 × 3 supercell was 1.75 and 7.44 Å for the ‘Str. 1’, and ‘Str. 2’, respectively. The DOS plot calculated for both the structures is shown in Fig. S2,† which indicates that the nature of band structure remains independent of lowering the dopant concentration. However, the band gap was found to strongly depend on the distance of separation between N and W. The calculated band gap for ‘Str. 1’ and ‘Str. 2’ was 3.12, and 2.36 eV, respectively. The interesting point is that the band gap was found to decrease upon reducing the dopant concentration (for ‘Str. 2’), while the reverse trend is much more common in practical scenarios. This can be achieved only by choosing an appropriate synthetic strategy.
3.5. Optical property
To observe the change in the optical spectrum of KNbO3 due to (N, W) codoping, frequency dependent dielectric function calculations were carried our employing the HSE hybrid functional. The dielectric function can be expressed in combination of real, ε1(ω) and imaginary, ε2(ω) parts as ε(ω) = ε1(ω) + iε2(ω). Herein, ε2(ω) is calculated by a summation over a sufficiently large number of unoccupied states, and then ε1(ω) from Kramer–Kronig relation, as implemented in VASP 5.2 version. The absorption coefficient, α(ω), can be calculated using these two parts of the dielectric function as follows:| |
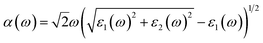 | (6) |
Fig. 8 shows the calculated absorption coefficient as a function of light wavelength. The absorption curve corresponding to undoped KNbO3 was found to be limited to the UV region. This is consistent with the calculation of the electronic band gap for KNbO3 (3.24 eV). Upon codoping the absorption spectrum was found to be shifted towards the visible region. This indicates that the (N, W)-codoped KNbO3 should have better visible light activity than the undoped KNbO3.
 |
| | Fig. 8 Optical spectrum for undoped KNbO3 and (N, W)-codoped KNbO3 (N: 4.17%, W: 12.5%). | |
3.6. Band edge alignment
To date, we have discussed how the band gap can be reduced to utilize the solar spectrum. However, this band gap narrowing may sometimes deactivate the oxidising or reducing behaviour of the parent material. To check whether these modified materials are suitable for overall water splitting or not, their band edge positions were aligned with respect to water redox levels. To be suitable for hydrogen production from water, the CBM of the material must be located above the H+/H2 level, whereas for oxygen generation, its VBM must be located below the H2O/O2 level. For example, for KNbO3, the CBM and VBM are located at around 0.78 eV above and 1.23 eV below from the respective water redox levels. Therefore, KNbO3 is active for overall water splitting, which was evident in several experimental studies. Because the VBM and CBM obtained in DFT calculation are not on an absolute scale, one cannot directly align their band edges with respect to water redox levels. For this purpose, we first fixed the location of VBM and CBM for the undoped KNbO3 based on the experimental data.54 Then, the band edges of the modified materials are aligned by calculating the relative shift of the VBM and CBM with respect to that of undoped KNbO3. Among the different configurations for the (N, W)-codoped system, we are interested in only those structures (‘Str. 2’ in each case) that have a lower band gap than the undoped KNbO3. Fig. 9 shows the CBM and VBM positions of the (N, W)-codoped KNbO3, with a different dopant concentration. The CBM was found to be 0.83, 0.52, and 0.6 eV above the H+/H2 level for the (N, W)-codoped KNbO3 with W-concentration corresponding to 12.5%, 8.33%, and 5.55%, respectively. On the other hand, the VBM for the respective case lies 0.72, 0.64, and 0.55 eV below the H2O/O2 level. This indicates that the band edges are in favourable positions to release both hydrogen and oxygen. Therefore, the (N, W)-codoped KNbO3 is suitable for overall water splitting over a wide range of concentrations.
 |
| | Fig. 9 Band edge alignment of undoped KNbO3 and (N, W)-codoped KNbO3 with different dopant concentrations (N: 4.17%, W: 12.5%), (N: 2.78%, W: 8.33%), and (N: 1.85%, W: 5.55%) with respect to the water redox levels. | |
4. Conclusions
In this theoretical investigation, the aim was to predict an efficient strategy to improve the visible light photocatalytic activity of KNbO3. All the electronic structure calculations were carried out using DFT with the HSE hybrid functional to present a more realistic description. The present study reveals that the calculated band gap with a 0.2 Å−1 screening parameter and 36% of mixing of exact HF exchange shows the best match (3.23 eV) with the experimentally reported band gap (3.24 eV) of KNbO3. Herein, we chose the (N, W) dopant pair to reduce the band gap of KNbO3. Although both the elements individually are able to lower the band gap of KNbO3 by a significant extent, the formation of associated vacancy defects due to charge mismatch may accelerate electron–hole recombination. Another factor is the downward shifting of the CBM by a large extent, which is not desirable because of the possibility of lowering the hydrogen evolution rate. However, these disadvantages were overcome in the case of the codoped system due to the formation of a charge compensated system and mainly valence band controlled band gap narrowing. This is the unique characteristics of the (N, W) pair in this case. Moreover, N doping was found to be more favoured in presence of W. The calculation of formation energy for codoping under different synthetic conditions provides useful information for practical situations. In addition, the present study has also indicated a strong dependency of the synthetic approach to achieve efficient band gap narrowing with the (N, W) pair. Interestingly, the band gap reduction became more prominent when the dopant–dopant separation was increased. This lead to the achievement of a considerable band gap reduction even at low concentration. The choice of (N, W) pair was again justified by its ability to narrow the band gap in a controlled way, so that the codoped material can satisfy the thermodynamic criterion for overall water splitting.
Acknowledgements
We thank the BARC Computer Centre for providing the high performance parallel computing facility. We also thank Dr B. N. Jagatap for his encouragement and support. S. K. G. is thankful to Department of Atomic Energy and Department of Science and Technology for the Raja Ramanna and Fellowship J. C. Bose Fellowship, respectively. B. M. acknowledges Mrs Pampa Modak and Dr K. Srinivasu for their valuable support.
References
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- F. Fresno, R. Portela, S. Suárez and J. M. Coronado, J. Mater. Chem. A, 2014, 2, 2863–2884 CAS.
- K. Srinivasu, B. Modak and S. K. Ghosh, J. Phys. Chem. C, 2014, 118, 26479–26484 CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- M. D. Hernández-Alonso, F. Fresno, S. Suárez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 Search PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- P. Zhang, J. Zhang and J. Gong, Chem. Soc. Rev., 2014, 43, 4395–4422 RSC.
- B. Modak and S. K. Ghosh, J. Phys. Chem. C, 2015, 119, 7215–7224 CAS.
- B. Modak, K. Srinivasu and S. K. Ghosh, J. Phys. Chem. C, 2014, 118, 10711–10719 CAS.
- J. Shi and L. Guo, Prog. Nat. Sci., 2012, 22, 592–615 CrossRef.
- H. Hayashi, Y. Hakuta and Y. Kurata, J. Mater. Chem., 2004, 14, 2046–2051 RSC.
- H. Shi and Z. Zou, J. Phys. Chem. Solids, 2012, 73, 788–792 CrossRef CAS.
- J. W. Liu, G. Chen, Z. H. Li and Z. G. Zhang, Int. J. Hydrogen Energy, 2007, 32, 2269–2272 CrossRef CAS.
- T. Zhang, K. Zhao, J. Yu, J. Jin, Y. Qi, H. Li, X. Hou and G. Liu, Nanoscale, 2013, 5, 8375–8383 RSC.
- L. Yan, J. Zhang, X. Zhou, X. Wu, J. Lan, Y. Wang, G. Liu, J. Yu and L. Zhi, Int. J. Hydrogen Energy, 2013, 38, 3554–3561 CrossRef CAS.
- Q.-P. Ding, Y.-P. Yuan, X. Xiong, R.-P. Li, H.-B. Huang, Z.-S. Li, T. Yu, Z.-G. Zou and S.-G. Yang, J. Phys. Chem. C, 2008, 112, 18846–18848 CAS.
- T. Zhang, W. Lei, P. Liu, J. A. Rodriguez, J. Yu, Y. Qi, G. Liu and M. Liu, Chem. Sci., 2015, 6, 4118–4123 RSC.
- L. Jiang, Y. Qiu and Z. Yi, J. Mater. Chem. A, 2013, 1, 2878–2885 CAS.
- J. Lan, X. Zhou, G. Liu, J. Yu, J. Zhang, L. Zhi and G. Nie, Nanoscale, 2011, 3, 5161–5167 RSC.
- L. Yan, T. Zhang, W. Lei, Q. Xu, X. Zhou, P. Xu, Y. Wang and G. Liu, Catal. Today, 2014, 224, 140–146 CrossRef CAS.
- Z. Wang, H. Gu, Y. Hu, K. Yang, M. Hu, D. Zhou and J. Guan, CrystEngComm, 2010, 12, 3157–3162 RSC.
- G. H. Khorrami, A. Kompany and Z. A. Khorsand, Mod. Phys. Lett. B, 2014, 28, 1450224 CrossRef CAS.
- J.-j. Zhang, H. Zhou, J.-S. Chen, T. Tang and Y.-Z. Hao, Adv. Mater. Res., 2014, 873, 783–786 CrossRef.
- R. Balakrishnaiah, D. W. Kim, S. S. Yi, K. D. Kim, S. H. Kim, K. Jang, H. S. Lee and J. H. Jeong, ECS Trans., 2009, 16, 23–31 CrossRef CAS.
- Y. Guo, Y. Li, S. Li, L. Zhang, Y. Li and J. Wang, Energy, 2015, 82, 72–79 CrossRef CAS.
- J. Wang, X. Wang, Z. Cui, B. Liu and M. Cao, Phys. Chem. Chem. Phys., 2015, 17, 14185–14192 RSC.
- R. Wang, Y. Zhu, Y. Qiu, C.-F. Leung, J. He, G. Liu and T.-C. Lau, Chem. Eng. J., 2013, 226, 123–130 CrossRef CAS.
- M. V. Dozzi and E. Selli, J. Photochem. Photobiol., C, 2013, 14, 13–28 CrossRef CAS.
- H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483–5486 CrossRef CAS.
- Z. Lin, A. Orlov, R. M. Lambert and M. C. Payne, J. Phys. Chem. B, 2005, 109, 20948–20952 CrossRef CAS PubMed.
- M. Batzill, E. H. Morales and U. Diebold, Phys. Rev. Lett., 2006, 96, 026103 CrossRef PubMed.
- Y. Nakano, T. Morikawa, T. Ohwaki and Y. Taga, Chem. Phys., 2007, 339, 20–26 CrossRef CAS.
- B. Modak and S. K. Ghosh, J. Phys. Chem. C, 2015, 119, 23503–23514 CAS.
- B. Modak, K. Srinivasu and S. K. Ghosh, RSC Adv., 2014, 4, 45703–45709 RSC.
- B. Modak, K. Srinivasu and S. K. Ghosh, Phys. Chem. Chem. Phys., 2014, 16, 17116–17124 RSC.
- B. Modak and S. K. Ghosh, Chem. Phys. Lett., 2014, 613, 54–58 CrossRef CAS.
- B. Modak, K. Srinivasu and S. K. Ghosh, Phys. Chem. Chem. Phys., 2014, 16, 24527–24535 RSC.
- W.-J. Yin, H. Tang, S.-H. Wei, M. M. Al-Jassim, J. Turner and Y. Yan, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 045106 CrossRef.
- J. Paier, M. Marsman, K. Hummer, G. Kresse, I. C. Gerber and J. G. Ángyán, J. Chem. Phys., 2006, 124, 154709 CrossRef CAS PubMed.
- B. Modak and S. K. Ghosh, Phys. Chem. Chem. Phys., 2015, 17, 15274–15283 RSC.
- B. Modak and S. K. Ghosh, J. Phys. Chem. B, 2015, 119, 11089–11098 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Adv., 1976, 32, 751–767 CrossRef.
- W. Zhu, X. Qiu, V. Iancu, X.-Q. Chen, H. Pan, W. Wang, N. M. Dimitrijevic, T. Rajh, H. M. Meyer III and M. P. Paranthaman, et al., Phys. Rev. Lett., 2009, 103, 226401 CrossRef PubMed.
- A. Tkach, A. Almeida, J. Agostinho Moreira, J. Perez de la Cruz, Y. Romaguera-Barcelay and P. M. Vilarinho, Appl. Phys. Lett., 2012, 100, 192909 CrossRef.
- K. A. McDonnell, N. J. English, M. Rahman and D. P. Dowling, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 115306 CrossRef.
- M. Chiodi, C. P. Cheney, P. Vilmercati, E. Cavaliere, N. Mannella, H. H. Weitering and L. L. Gavioli, J. Phys. Chem. C, 2012, 116, 311–318 CAS.
- W.-J. Shi and S.-J. Xiong, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 205210 CrossRef.
- R. Long and N. J. English, Chem. Mater., 2010, 22, 1616–1623 CrossRef CAS.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figure for the 2 × 2 × 3 and 2 × 3 × 3 supercells of the KNbO3 crystal structure, indicating the dopant positions. Density of states plot for the (N, W)-codoped-KNbO3 using 2 × 2 × 3 and 2 × 3 × 3 supercells. See DOI: 10.1039/c5ra26079e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 









