
Fluorine substitution effects of halide anion receptors based on the combination of a distinct hydrogen bond and anion–π noncovalent interactions: a theoretical investigation†
 c
c
Abstract
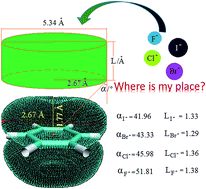
Noncovalent interactions between halide anions and a series of N-(4-vinyl-2-benzoic acid benzyl ester)-phenyl-urea containing receptors (1–8) based on hydrogen bond and (or) anion–π interactions were investigated via theoretical calculations based on dispersion corrected density functional B3LYP-D3. Particularly, the fluorine substitution effects were deeply explored. The results showed that the substituent number and position of fluorine groups on the phenyl ring of the benzoic acid esters group has a significant effect on the configuration and cooperative properties of the hydrogen bond and anion–π interactions. Consequently, a more feasible and rational geometric criterion for either a strong or weak halide-anion–π contact was proposed via three inequalities independent of any empirical parameters, which is different from the criterion proposed very recently by Albrecht and Rissanen based on their experience with solid state anion–π interactions (Chem. Sci., 2015, 6, 354–359). Additionally, electronic properties and behavior of the systems were discussed according to the calculations on the frontier molecular orbital, total electrostatic potential and visualized weak interactions regions. It is expected that the theoretical explorations from a molecular level in this work may be useful for future experimental study and helpful for understanding the structure–activity relationship between aromatic rings and anion–π interactions in the case of combination of distinct noncovalent interactions.
 Please wait while we load your content...
Please wait while we load your content...

