
Aminal linked inorganic–organic hybrid nanoporous materials (HNMs) for CO2 capture and H2 storage applications†
Raeesh Muhammad,
Pawan Rekha and
Paritosh Mohanty*
Department of Chemistry, Indian Institute of Technology Roorkee, Roorkee, Uttarakhand-247667, India. E-mail: pmfcy@iitr.ac.in; paritosh75@gmail.com
First published on 2nd February 2016
Abstract
Nitrogen-rich aminal linked inorganic–organic hybrid nanoporous materials (HNMs) with cyclophosphazene moieties in the frameworks were synthesized by a Schiff base condensation reaction. The ultra-microporous materials have a specific surface area (SBET) upto 976 m2 g−1 and could capture 18.9 wt% CO2 and 1.6 wt% H2 at 273 and 77 K, respectively, at 1 bar. The materials have a nitrogen content upto 42% which is the highest among the nanoporous materials category. The high nitrogen content is beneficial for several applications such as CO2 capture.
1. Introduction
The recent advancement in the synthesis of inorganic–organic hybrid nanoporous materials (HNMs) possessing high surface area, tuneable pore structure, low density, mechanical and hydrothermal stability with tailor-made functionalities have drawn tremendous interest among researchers in the areas of catalysis, drug delivery, environment, gas sorption and separation applications.1–3 In general, the inorganic moiety provides the thermal stability, mechanical strength, and structural order, whereas, the tuneable functionality, porosity, hydrophobicity, optical and electrical properties of the organic moiety make hybrid materials superior to their both inorganic and organic counter parts.4 Achieving permanent porosity has always been a challenge, as nature tends to have a compact structure with minimum volume having least energy and surface area.5 Recently, our group has reported the synthesis of cyclophosphazene based inorganic–organic hybrid porous materials.6–8 It was observed that the cyclophosphazene units in the framework has the synergic effect in achieving the high surface area in the obtained hybrid materials. This was mainly due to the paddle-wheel structure of the cyclophosphazene derivatives as reported earlier.6–11The classical Schiff base condensation discovered by Hugo Schiff12 in 1864 has traditionally been used for the generation of imine and aminal linkages by condensing aldehydes with different amines.13,14 In general, the imine double bond is formed, however, primary amines may subsequently attack resulting exclusively aminal linkages.15 The mechanism was thoroughly investigated and involves a number of reversible steps. The formation of the imine and aminal linkages have been exploited to prepare various types of organic polymers.16,17 Moreover, recently the reaction has been used for the synthesis of nanoporous organic polymers with high surface area having imine and aminal linkages.18–24 To the best of our knowledge, there is no report available for the synthesis of nanoporous inorganic–organic hybrid materials using Schiff base condensation. In this paper, we report the synthesis of aminal linked inorganic–organic hybrid nanoporous materials using Schiff base condensation.
The first step involves the synthesis of aldehyde precursor (compound-I) and the second step involves the condensation of the compound-I with melamine as shown in Scheme 1. Melamine was chosen in this research to facilitate the condensation and most importantly the synthesized material will have larger nitrogen content of 40% or more, which has not been achieved so far for any types of inorganic–organic hybrid materials. The best reported nitrogen content for any nanoporous material was 40.42% for SNW-3.20 The larger nitrogen content in the material will be very much beneficial for the gas sorption applications, as the lone pair of electrons in the nitrogen acts as Lewis base and potentially could interact with Lewis acidic gases such as CO2. The capture and separation of CO2 from the flue gas stream has been considered to be the potential solution for global warming.25–27 Moreover, the hybrid materials with ultra-micropores could be exploited for the hydrogen storage applications. Hydrogen is being considered an ideal fuel for future given its abundance and non polluting nature when used with fuel cells, and it has emerged as an ideal substitute for fossil fuels.28–34 Finding the suitable adsorbents for both CO2 capture and H2 storage is a great challenge and leads to the extensive research in the recent times.
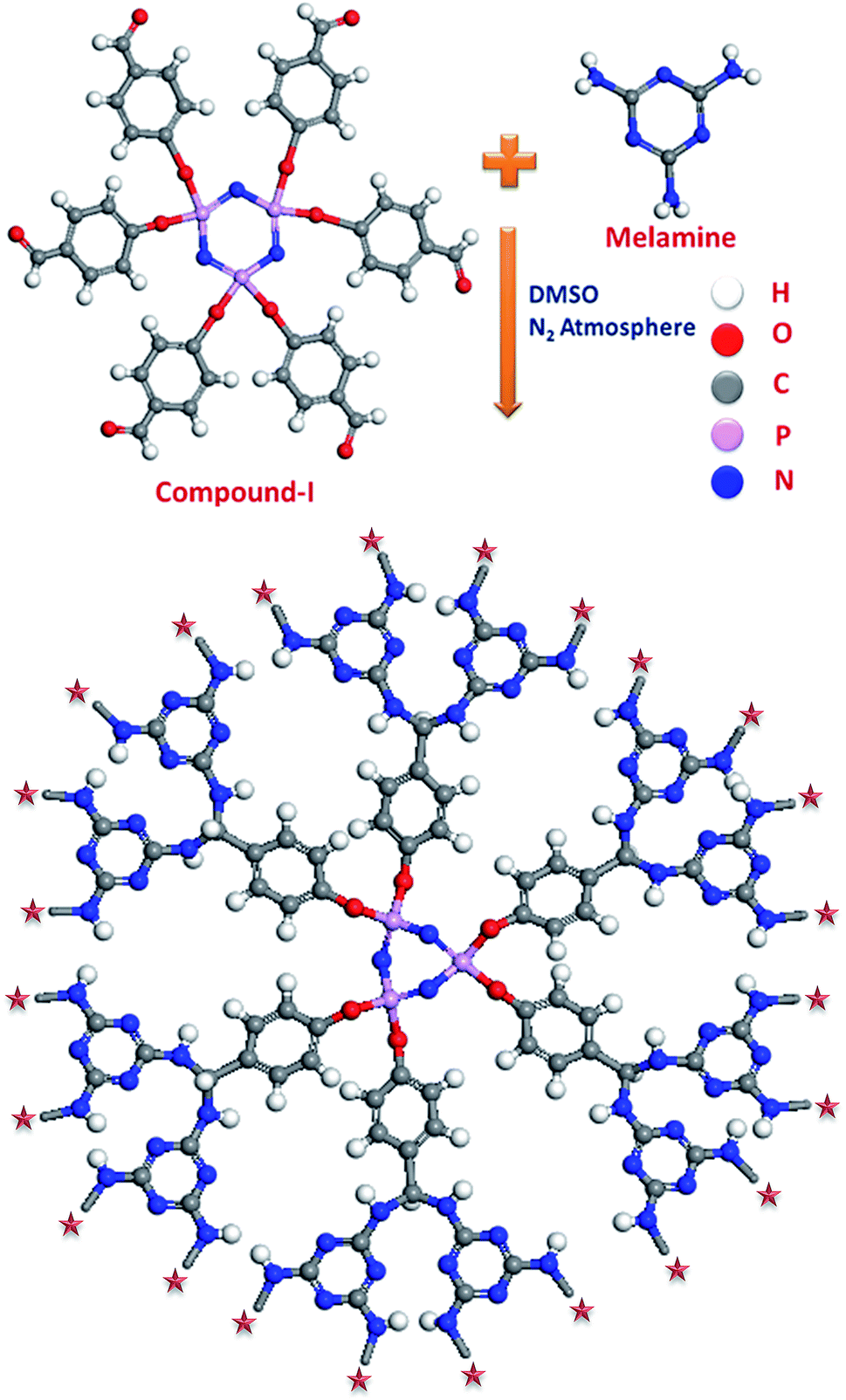 | ||
| Scheme 1 Reaction scheme for the synthesis of HNMs. | ||
2. Experimental section
2.1 Materials
Phosphonitrilic chloride trimer (PNC) (99%, Sigma Aldrich, India), melamine (99%, Sigma Aldrich, India), p-hydroxybenzaldehyde (99%, Sigma Aldrich, India), dimethylsulfoxide (DMSO) (Fisher Scientific, India), Na2SO4 (Fisher Scientific, India), K2CO3 (Fisher Scientific, India), tetrahydrofuran (THF) and dichloromethane (DCM) (Fisher Scientific, India) were of analytical grade and used as received.2.2 Synthesis of compound I
The compound-I was synthesised by following the work of Kagit et al. with some modification as shown in Scheme S1.†9 Typically, the solution of PNC 3.48 g, (10.00 mmol) in dry THF (50 ml, dried by passing over anhydrous Na2SO4) was added to a magnetically stirred solution of p-hydroxybenzaldehyde (7.46 g, 61.00 mmol) and K2CO3 (39.75 g, 121 mmol) in dry THF (100 ml) under N2 atmosphere. The reaction mixture was stirred for 48 h at RT. After this insoluble salts were removed by filtration and the soluble product in THF was obtained by removing THF under reduced pressure. It was then re-dissolved in DCM and further combined phase of DCM was extracted with water and brine solution (50 ml each). The extraction was repeated for three times. The organic phase was dried by passing it through anhydrous Na2SO4. The final product was obtained by removing the DCM under reduced pressure which gives white crystal on recrystallization with ethylacetate. The detailed characterization of compound-I is given in Fig. S1–S4.†2.3 Synthesis of HNMs
The HNMs have been synthesized, as shown in Schemes 1 and S2 (ESI†), by Schiff base condensation of compound-I with melamine. Typically, 1.5 mmol (1.293 g) of compound-I dissolved in 5 ml of DMSO was added drop-wise to a solution of 4.5 mmol (0.567 g) of melamine in 25 ml of DMSO under N2 atmosphere with continuous stirring. The temperature was then raised to 453 K for 48 h under stirring condition. A off-white precipitate was formed which was filtered and washed with acetone, methanol and finally soxhlet extracted with diethyl ether for 8 h to exchange the DMSO from the pores of the framework. The product was dried under vacuum at 373 K for 18 h to give the final product designated as HNM-1.2.4 Characterization
FT-IR analysis was carried out by using PerkinElmer Spectrum Two. The 31P and 13C cross polarization magic angle spinning (CP-MAS) NMR spectra were recorded on JEOL resonance JNM-ECX-400II at 161.83 MHz (31P), and 100 MHz (13C) with sample spinning frequency of 6 and 10 kHz, respectively. Total number of scans for 31P and 13C were 512 and 9000, respectively. X-ray diffraction patterns were obtained using a Rigaku Ultima IV with Cu-Kα radiation source (λ = 0.15405 nm) with a scanning speed of 5° per min measured in the range of 10 to 80° in the 2θ scale. The microstructural analysis of the samples was done using the instrument “TESCAN MIRA 3” and the samples were measured at accelerating voltage of 10 kV. Before the analysis, the samples were dispersed in sticky carbon tape attached to a flat aluminium sample holder. Then samples were coated with gold by a standard sputtering technique for 60 s at 1 × 10−4 mbar of pressure in N2 atmosphere before imaging. TEM images were recorded by using TECNAIG2S-TWIN with field emission gun operating at 200 kV. The samples were prepared by dispersing in ethanol using sonicator and dropping it over carbon coated copper grid. The elemental analysis (C/H/N/S/O) was performed using Thermo Flash 2000. The elemental composition of nitrogen (N), carbon (C), hydrogen (H) and oxygen (O) was determined by thermal conductivity detector (TCD). The amount of sample loaded in the sample vial was 2–3 mg. Thermogravimetric analysis was carried out under air with a heating rate of 5 K min−1 at a flow rate of 200 ml min−1 with alumina powder as reference using the instrument “EXSTAR TG/DTA6300”.Porosity and gas sorption studies were performed using Autosorb iQ2 volumetric physisorption analyzer (Quantachrome Instruments, USA) using adsorbate of ultra-high purity (99.999%, Sigma Gases and Services, India). The N2 sorption was performed at 77 K using liquid nitrogen bath after degassing the specimen at 393 K for 6 h with heating rate of 5 K min−1. The specific surface area was calculated by fitting the gas sorption data to BET and Langmuir model in the relative pressure range of 0.05 to 0.30. Pore size distribution was calculated by fitting the nitrogen sorption isotherm to density functional theory (DFT) model using Kernel: “N2 at 77 K on carbon (slit pore, QSDFT equilibrium model)”. CO2 and CH4 adsorption isotherms were measured at 273 and 298 K using chiller/circulator. The temperature of chiller/circulator bath was maintained by using the mixture of ethylene glycol and water in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volumetrically. The hydrogen adsorption isotherms were measured at 77 K using liquid nitrogen bath. The isosteric heat of adsorption (Qst) for CO2 and CH4 was calculated using the Clausius–Clapeyron equation.
:1 volumetrically. The hydrogen adsorption isotherms were measured at 77 K using liquid nitrogen bath. The isosteric heat of adsorption (Qst) for CO2 and CH4 was calculated using the Clausius–Clapeyron equation.
3. Results and discussion
In order to confirm the structure as proposed in Scheme 1, the HNM-1 has been investigated by FT-IR and 31P, 13C CP-MAS NMR spectroscopy. The broad bands at 3405 and 1650 cm−1 in the FT-IR spectrum of the HNM-1 as shown in Fig. 1a and Table S1, (ESI†) due to the (–N–H) stretching and bending vibration, respectively, indicate the formation of the aminal linkage.20,21 The formation of the aminal linkage was supported by the absence of the characteristic imine band around 1620 cm−1 due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration.20,21 The band at 2930 cm−1 was assigned to –C–H stretching of tertiary carbon due to the aminal linkage.20 The band at 1548 and 1477 cm−1 were attributed to triazine moiety of melamine.20 The absence of the CO stretching vibration around 1700 cm−1 confirms the complete condensation of compound-I with melamine.20,21 The observation of bands in the range of 1203 to 1193, 986 and 584 cm−1 assigned to νas(PN–P), νas(P–O–P) and δ(PN–P) vibrations, respectively, confirm the inclusion of the cyclophosphazene units in the frameworks.6–10
N stretching vibration.20,21 The band at 2930 cm−1 was assigned to –C–H stretching of tertiary carbon due to the aminal linkage.20 The band at 1548 and 1477 cm−1 were attributed to triazine moiety of melamine.20 The absence of the CO stretching vibration around 1700 cm−1 confirms the complete condensation of compound-I with melamine.20,21 The observation of bands in the range of 1203 to 1193, 986 and 584 cm−1 assigned to νas(PN–P), νas(P–O–P) and δ(PN–P) vibrations, respectively, confirm the inclusion of the cyclophosphazene units in the frameworks.6–10
 | ||
| Fig. 1 (a) FT-IR spectrum, (b) 13C CP-MAS NMR spectrum and (c) 31P CP-MAS NMR spectrum of HNM-1. | ||
The detailed analysis about the chemical structure of HNM-1 was performed by 13C and 31P CP-MAS NMR spectroscopy. The 13C CP-MAS NMR spectrum (Fig. 1b) shows the resonance signals at 166, 155, 135, 128, 122 and 57 ppm corresponding to carbon no. 1, 2, 3, 4, 5 and 6, respectively (Fig. 1b, Table S2, ESI†).20 The absence of signal at 160 ppm (due to imine linkage) and the observation of the signal at 57 ppm (due to the tertiary carbon) confirm the formation of the aminal linkages in the HNM-1.20 The observation of a single resonance signal at 9.6 ppm in the 31P CP-MAS spectrum of HNM-1 (Fig. 1c) confirms the incorporation of the cyclophosphazene units in the framework.7–9
The aminal linkage in the HNM-1 was further supported by the elemental analysis (Table S3, ESI†). Assuming a fully condensed form having aminal linkage as shown in Scheme 1, the theoretical composition of the HNM-1 would be C78H66N75O3P3. Thus, it will have 41.77, 46.84, 2.97, and 4.28% of C, N, H and O, respectively. The observed elemental composition for HNM-1 was 40.18, 42.16, 4.46 and 5.25% for C, N, H and O, respectively, which was fairly matching with the theoretical values. The excess O and H observed were mainly due to the terminal functionality and trapped moisture in the nanopores. Further, assuming the imine linkage in HNM-1, the theoretical composition would be C60H30N39O6P3 with 48.49, 36.76, 2.03 and 6.46% of C, N, H and O, respectively, which was not matching with the experimental results.
The HNM-1 was thermally stable up to a temperature of 623 K in air as evaluated by the TGA/DTG analysis (Fig. S5, ESI†). The HNM-1 was further found to be X-ray amorphous (Fig. S6, ESI†). The particles of size 20 to 50 nm forming the network could be seen from the FESEM and TEM images (Fig. 2). The inter-particulate pores could be seen in the FESEM image (Fig. 2a), however, nanoporous nature of the HNM-1 can be clearly seen with the TEM image (Fig. 2b). The SAED pattern in the inset of Fig. 2b further confirms the amorphous nature of the specimen.
 | ||
| Fig. 2 (a) FESEM and (b) TEM image of HNM-1. SAED pattern in the inset of (b). | ||
The formation of the aminal frameworks as proposed in the Scheme 1, and the observation of the nanoporous structure in the FESEM and TEM images, has encouraged us to study the textural properties of the HNM-1. Fig. 3a shows the N2 sorption isotherm of HNM-1 measured at 77 K. A type-I isotherm with steep N2 uptake at low pressure (below P/P0 = 0.01) and significant multilayer adsorption in intermediate section with narrow hysteresis in the higher pressure range could be seen. The hysteresis at higher pressure could be due to significant N2 adsorption between the particles of very small size (20–50 nm) and due to the external surface area in the interparticulate voids. The steep N2 uptake at low pressure indicates the presence of ultra-micropores in the specimen with minor mesopores and macropores. The pore size distribution (PSD) estimated by DFT model (Fig. S9†) shows the average pore diameter of 0.65 nm and majority of the pores were <2 nm in diameters, with some distribution in the higher diameter range. The specific surface area calculated by using BET (SBET) and Langmuir (SLang) model were found to be to 976 and 1237 m2 g−1, respectively (Fig. S10†). These values are further comparable with the cumulative surface area (967 m2 g−1) calculated by DFT & Monte-Carlo analysis. The multilayer adsorption in intermediate section of N2 isotherm indicated the presence of significant external surface area. The external surface area calculated by the t-plot method was found to be 603 m2 g−1. This further supports the smaller particle size of the sample. Pore volume calculated at relative pressure (P/P0) 0.90 from the adsorption branch of the isotherm, was found to be 1.07 cm3 g−1 (Table S4, ESI†). The higher specific surface area and pore volume of HNM-1 could be attributed to the unique paddle wheel structure of the cyclophosphazene moieties in the framework, which has been reported earlier.6–8,10 In order to further understand if the reaction temperature has any profound effect on the textural properties, two more samples have been synthesized at 423 and 393 K (while keeping all other reaction conditions same) designated as HNM-2 and HNM-3, respectively. It was observed that with the decrease in the reaction temperature, there was the decrease in the SBET to 807 and 513 m2 g−1, in HNM-2 and HNM-3, respectively (Fig. S7, S11–S12, Table S4, ESI†). The pore size distribution calculated by DFT model for HNM-2 and HNM-3 was found to be centred at about 0.64 nm. This could be attributed to the weaker framework condensation at lower temperature. High temperature led to opening of micropore that led to increase of both micropore as well as external surface area of HNM-1 in comparison to HNM-2 and HNM-3 (pl. see ESI, Table S4†). However, there was no substantial difference in the particle size and shape (Fig. S8†) although, the experiments were carried out at three different temperatures. The presence of the ultra-micropores with high SBET and pore volume coupled with the high nitrogen content (>42%) encouraged us to study the CO2 sorption properties. Fig. 3b shows the CO2 sorption isotherms of HNM-1 measured at 273 and 298 K. Almost complete reversibility was observed with minor hysteresis. The highest CO2 uptake was found to be 18.9 and 12.3 wt% at 273 and 298 K, respectively (Table S5, ESI†). The CO2 uptake of HNM-1 is higher than many of the recently reported materials like TBILP-1,35 PAF-336 and C-NP37 (Table S6, ESI†). The HNM-2 and HNM-3 also show noteworthy CO2 uptake of 13.2 wt% and 9.5 wt%, respectively at 273 K and 1 bar (Fig. S13 and Table S5†). It is, however, important to note that the high nitrogen content is not the only factor that can tune the CO2 capture capacity. There could be multiple factors that affect the CO2 capture capacity such as pore size, pore geometries, surface functionalities and kinetics. The role of high nitrogen content is to enhance the Lewis basic nature of framework which helps in the adsorption of Lewis acidic gas like CO2. The higher CO2 uptake of HNM-1 in comparison to HNM-2 and HNM-3 could be due to higher surface area and pore volume.
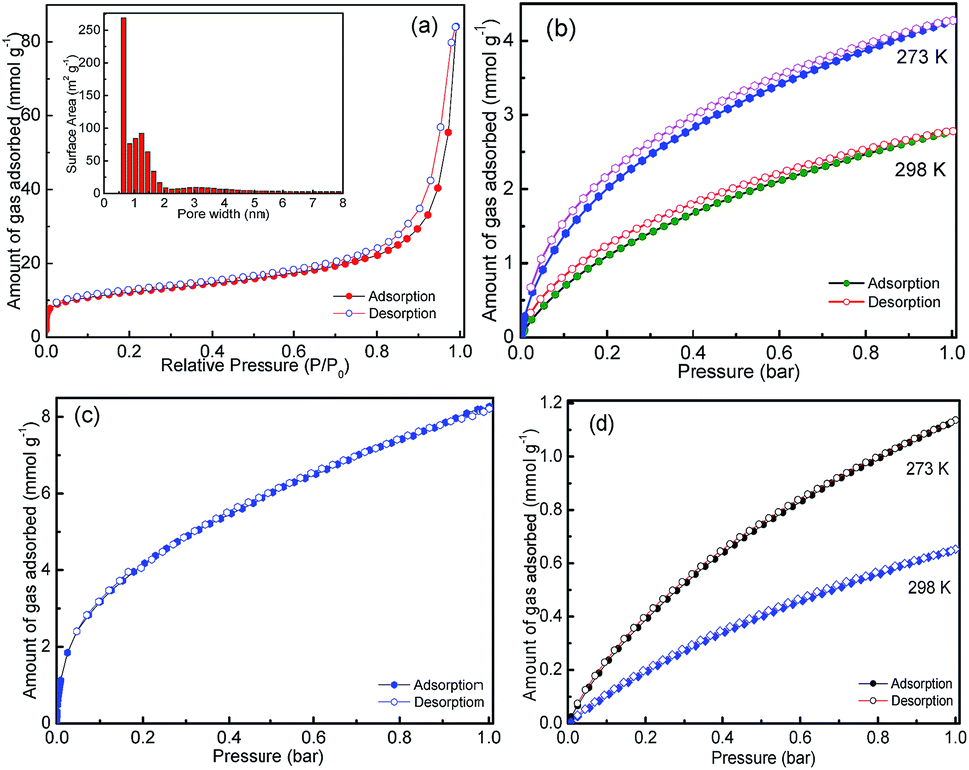 | ||
| Fig. 3 Gas sorption isotherms of HNM-1. (a) N2 sorption isotherm measured at 77 K, (b) CO2 sorption isotherms measured at 273 and 298 K, (c) H2 sorption isotherm measured at 77 K and (d) CH4 sorption isotherms measured at 273 and 298 K. The pore size distribution histogram calculated by the DFT model from the N2 sorption data is given in the inset of (a). | ||
The isosteric heats of adsorption (Qst) of HNMs were further calculated using Clausius–Clapeyron equation (Fig. S14, ESI†). At the onset, the Qst values are 34.7, 35.9 and 33 kJ mol−1 for HNM-1, HNM-2 and HNM-3, respectively (Table S5, ESI†). The Qst values of HNMs indicated the interaction between the adsorbent and the adsorbate was neither purely physisorption nor pure chemisorption and was at the cusp of these two.10
Furthermore, HNMs were tested for the H2 and CH4 storage. The H2 sorption isotherm as shown in Fig. 3c was completely reversible without hysteresis and the uptake at maximum pressure range for the HNM-1 was 1.63 wt% at 77 K and 1 bar. It is further worth mentioning that the sorption isotherm was not saturated in the measured pressure range indicating a higher uptake could be achieved at higher pressure.38 The H2 uptake of HNM-1 surpasses or comparable to various porous organic polymers and nanoporous hybrid materials such as, PSN-3,31 CTC-COF,32 PIMs33 and BLP(2H).34 The CH4 uptake of HNM-1 was found to be 1.13 and 0.88 wt% measured at 273 and 298 K, respectively. To understand the feasible application of the synthesized materials for the purpose of post combustion CO2 capture in coal fired power plants, the selective uptake of CO2 over CH4 and N2 is one of the main criteria. The selectivity of CO2 with respect to CH4 and N2 was estimated by using the Henry's law constant.10 The selectivity of CO2 over N2 and CO2 over CH4 for HNM-1 were found to be 62 and 14, respectively, at 273 K (Fig. S16–S21 and Table S5, ESI†). The selective uptake of HNMs is comparable to the recently reported materials like TBILP-1,35 PECONF-310 and higher than TBILP-2,35 C-NP,37 PAN-1,39 and fl-CTF35040 (Table S6, ESI†). The moderate selectivity shown by HNMs despite having very high nitrogen content could be attributed to the fact that CO2 might not be interacting with all the nitrogen of the framework and it is interacting with only nitrogen present on the surface.
4. Conclusions
In summary, Schiff base condensation has been used for the first time to synthesize aminal linked nanoporous inorganic–organic hybrid materials, which shows relatively high specific surface area and could capture up to 18.9 wt% of CO2 at 273 K along with 1.63 wt% of H2 at 77 K. The materials synthesized in this work has a higher nitrogen content (>42%) which is beneficial for exploiting the Lewis basic character needed for several applications. Further, the synthesis method could be extended to make various other cyclophosphazene derived nanoporous hybrid materials.Acknowledgements
The work was financially supported by DST, Govt. of India with Grant code DST/IS-STAC/CO2-SR-132/12(G).Notes and references
- C. Sanchez, P. Belleville, M. Popall and L. Nicole, Chem. Soc. Rev., 2011, 40, 696–753 RSC.
- D. Wang, W. Yang, S. Feng and H. Liu, Polym. Chem., 2014, 5, 3634–3642 RSC.
- L. Nicole, C. L. Robert, L. Rozes and C. Sanchez, Nanoscale, 2014, 6, 6267–6292 RSC.
- M. H. Lim and A. Stein, Chem. Mater., 1999, 11, 3285–3295 CrossRef CAS.
- P. Pierce, Structure in Nature is a Strategy for Design, MIT Press, 1990 Search PubMed.
- P. Rekha, U. Sahoo and P. Mohanty, RSC Adv., 2014, 4, 34860–34863 RSC.
- P. Rekha, R. Muhammad and P. Mohanty, RSC Adv., 2015, 5, 67690–67699 RSC.
- P. Rekha, V. Sharma and P. Mohanty, Microporous Mesoporous Mater., 2016, 219, 93–102 CrossRef CAS.
- R. Kagit, M. Yildirim, O. Ozay, S. Yesilot and H. Ozay, Inorg. Chem., 2014, 53, 2144–2151 CrossRef CAS PubMed.
- P. Mohanty, L. D. Kull and K. Landskron, Nat. Commun., 2011, 2, 401–406 CrossRef PubMed.
- H. R. Allcock, Chemistry and Applications of Polyphosphazenes, Wiley, New Jersey, 2002 Search PubMed.
- H. Schiff, Justus Liebigs Ann. Chem., 1864, 131, 118–119 CrossRef.
- N. E. Borisova, M. D. Reshetova and Y. A. Ustynyuk, Chem. Rev., 2007, 107, 46–79 CrossRef CAS PubMed.
- L. S. M. Forlani and P. E. Todesco, Gazz. Chim. Ital., 1986, 116, 229–232 CAS.
- S. J. Rowan and J. F. Stoddart, Org. Lett., 1999, 1, 1913–1916 CrossRef CAS.
- M. Y. Khuhawar, M. A. Mughal and A. H. Channar, Eur. Polym. J., 2004, 40, 805–809 CrossRef CAS.
- Y. Xin and J. Yuan, Polym. Chem., 2012, 3, 3045–3055 RSC.
- F. J. U. Romo, J. R. Hunt, H. Furukawa, C. Klock, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef PubMed.
- P. Pandey, A. P. Katsoulidis, I. Eryazici, Y. Wu, M. G. Kanatzidis and S. T. Nguyen, Chem. Mater., 2010, 22, 4974–4979 CrossRef CAS.
- M. G. Schwab, B. Fassbender, H. W. Spiess, A. Thomas, X. Feng and K. Mullen, J. Am. Chem. Soc., 2009, 131, 7216–7217 CrossRef CAS PubMed.
- W. C. Song, X. K. Xu, Q. Chen, Z. Z. Zhuang and X. H. Bu, Polym. Chem., 2013, 4, 4690–4696 RSC.
- A. Laybourn, R. Dawson, R. Clowes, J. A. Iggo, A. I. Cooper, Y. Z. Khimyak and D. J. Adams, Polym. Chem., 2012, 3, 533–537 RSC.
- Y. Zhu, H. Long and W. Zhang, Chem. Mater., 2013, 25, 1630–1635 CrossRef CAS.
- R. Gomes, P. Bhanja and A. Bhaumik, Chem. Commun., 2015, 51, 10050–10053 RSC.
- D. Lee, C. Zhang, C. Wei, B. L. Ashfeld and H. Gao, J. Mater. Chem. A, 2013, 1, 14862–14867 CAS.
- S. D. Kenarsari, D. Yang, G. Jiang, S. Zhang, J. Wang, A. G. Russell, Q. Wei and M. Fan, RSC Adv., 2013, 3, 22739–22773 RSC.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna, Z. Wei and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 7480–7484 CrossRef CAS PubMed.
- G. Li, B. Zhang, J. Yan and Z. Wang, Chem. Commun., 2014, 50, 1897–1899 RSC.
- N. B. McKeown, P. M. Budd and D. Book, Macromol. Rapid Commun., 2007, 28, 995–1002 CrossRef CAS.
- V. Sharma, A. Sahoo, Y. Sharma and P. Mohanty, RSC Adv., 2015, 5, 45749–45754 RSC.
- G. Li, B. Zhang and Z. Wang, Macromol. Rapid Commun., 2014, 35, 971–975 CrossRef CAS PubMed.
- J. T. Yu, Z. Chen, J. L. Sun, Z. T. Huang and Q. Y. Zheng, J. Mater. Chem., 2012, 22, 5369–5373 RSC.
- B. S. Ghanem, K. J. Msayib, N. B. McKeown, K. D. M. Harris, Z. G. Pan, P. M. Budd, A. Butler, J. Selbie, D. Book and A. Walton, Chem. Commun., 2007, 43, 67–69 RSC.
- K. T. Jackson, T. E. Reich and H. M. El-Kaderi, Chem. Commun., 2012, 48, 8823–8825 RSC.
- A. K. Sekizkardes, S. Altarawneh, Z. Kahveci, T. İslamoğlu and H. M. El-Kaderi, Macromolecules, 2014, 47, 8328–8334 CrossRef CAS.
- T. Ben, C. Pei, D. Zhang, J. Xu, F. Deng, X. Jinga and S. Qiu, Energy Environ. Sci., 2011, 4, 3991–3999 CAS.
- M. Saleh and K. S. Kim, RSC Adv., 2015, 5, 41745–41750 RSC.
- B. Adeniran and R. Mokaya, J. Mater. Chem. A, 2015, 3, 5148–5161 CAS.
- G. Li, B. Zhang, J. Yan and Z. Wang, Macromolecules, 2014, 47, 6664–6670 CrossRef CAS.
- S. Hug, M. B. Mesch, H. Oh, N. Popp, M. Hirscher, J. Senker and B. V. Lotsch, J. Mater. Chem. A, 2014, 2, 5928–5936 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, XRD, TGA, multi-point BET plot, Langmuir plot, selectivity and isosteric heat of adsorption. See DOI: 10.1039/c5ra25933a |
| This journal is © The Royal Society of Chemistry 2016 |