
Superparamagnetic nalidixic acid grafted magnetite (Fe3O4/NA) for rapid and efficient mercury removal from water†
Syed M. Husnaina,
Jae-Hawn Kima,
Chung-Seop Lee a,
Yoon-Young Changb,
Wooyong Umcd and
Yoon-Seok Chang*a
a,
Yoon-Young Changb,
Wooyong Umcd and
Yoon-Seok Chang*a
aSchool of Environmental Science and Engineering, Pohang University of Science and Technology (POSTECH), Pohang 790-784, Republic of Korea. E-mail: yschang@postech.ac.kr
bDepartment of Environmental Engineering, Kwangwoon University, Seoul 139-701, Republic of Korea
cDivision of Advanced Nuclear Engineering, POSTECH, Republic of Korea
dPacific Northwest National Laboratory, Richland, USA
First published on 5th April 2016
Abstract
A new nanomaterial, nalidixic acid grafted magnetite (Fe3O4/NA), was synthesized via a chemical reaction with nano sized magnetite particles. The Fe3O4/NA was superparamagnetic at room temperature and could be separated by an external magnetic field. The presence of mercury in groundwater in wide scale industrial areas of the world has been a huge problem and the prepared Fe3O4/NA nanoparticles showed a high adsorption capacity towards Hg(II) as compared to bare magnetite particles. The high adsorption capacity of NA grafted Fe3O4 (9.52 mg g−1) was due to the increased adsorption sites in the magnetite-nalidixic acid (Fe3O4/NA). The sorption equilibrium data obeyed the Langmuir model while kinetic studies demonstrated that the sorption process of Hg(II) followed well the pseudo second order model. Since the Fe3O4/NA showed (over 99.8%) removal of the initial 1000 ppb Hg(II) within 60 min, it should be practically usable for Hg(II) contaminated water. The desorption of Hg(II) loaded on Fe3O4/NA could be successfully achieved with 0.001 M HCl containing 0.3 M thiourea, and the sorbent exhibited excellent reusability.
Introduction
Toxic, heavy metal contamination in water has remained a matter of great environmental concern for the last few years. In the context of toxic metals, mercury is considered to be one of the most poisonous metals, widely found in surface and groundwater.1,2 Owing to its high bioaccumulation and biomagnification potential, the presence of minute concentrations in water causes severe problems to both aquatic organisms and human beings.3 Exposure of mercury may provoke a variety of detrimental health effects of the central nervous system, gastrointestinal tract, liver, impairment of pulmonary, kidney function and chest pains.4,5 Many cases of Minamata disease were diagnosed among the general population due to consumption of fish and shellfish in different countries around the world.6 Major contamination sources through which mercury enters the water bodies are through wastes from industrial processes such as chloralkali, paper and pulp, nuclear fuel production, mining, electroplating, oil refinery, paint and battery manufacturing, and etc.3,7 As a result of its toxic nature, the US Environmental Protection Agency (US EPA) has prescribed the maximum level of mercury contamination in drinking water at 2 ppb.8The removal of aqueous mercury can be achieved by variety of conventional techniques including coagulation,7 precipitation,9 ion exchange,10 membrane separation,11 reduction,12 amalgamation,13 and adsorption.14 Among these, adsorption is considered to be the most effective and simplest approach due to its low cost, ease of operational setup, and availability of wide range of adsorbents.15,16
In the recent years, magnetic nanoparticles (MNPs) have been extensively used in water treatment because of their cost-effectiveness, extremely small size, high surface to volume ratio and convenient magnetic field assisted separation.17 However, it has been observed that bare magnetic nanoparticles are highly susceptible to auto oxidation, easily agglomerated and not target selective in various environmental conditions.18,19 These factors can be compensated with help of surface coating using inorganic materials,20,21 polymers,22,23 carbon,24,25 and biomolecules26,27 to show high stability and enhanced sorption capacity.
Active Pharmaceutical Ingredients (APIs), one of major classes of organic compounds, have been investigated as metal extracting agents for radioactive, transition and rare earth metals.28 Among them, nalidixic acid (NA shown in Fig. 1) was used in fluorometric determination of some lanthanides,29 spectrophotometric determination of Fe(III),30 online pre-concentration of uranium and scandium on XAD-4 resin,31,32 and liquid–liquid extraction of Eu(III) and Nd(III) in dichloromethane.33 Remediation of heavy metals via NA or magnetite based composites has motivated us to synthesize magnetite-nalidixic acid (Fe3O4/NA) nanoparticles for Hg(II) removal.
 | ||
| Fig. 1 Chemical structure of nalidixic acid. | ||
Here, we report a synthesis of novel kind of magnetite grafted with NA in situ at low temperature (80 °C). The prepared Fe3O4/NA showed nearly complete removal (99.8%) of Hg(II) below 2 ppb in water, as a practical approach for Hg(II) removal from water due to their increased sorption sites in the presence of NA.
Experimental
Materials
The chemicals used in these experiments were of analytical grade and used as received. Nalidixic acid was procured from Sigma Aldrich (98% purity). Stock solutions were prepared by dissolving salts in Milli-Q water.Preparation of Fe3O4/NA
Fe3O4/NA was synthesized by chemical co-precipitation method. The procedure is shown schematically in Fig. 2. First 2.85 g of FeCl3·6H2O and 1.35 g of FeSO4·7H2O were dissolved in 50 mL of Milli-Q water and heated to 80 °C on the hot plate. Afterward, 43 mg of NA solid and 6 mL of ammonium hydroxide (25%) solution were rapidly and sequentially added into the vessel. The mixture was continually stirred at 80 °C for 40 min and then cooled to room temperature. The black Fe3O4/NA precipitates were collected by using an external magnet, rinsed several times with Milli-Q water, and dried in a vacuum oven. The control sample of bare Fe3O4 MNPs was also prepared in a similar way without NA addition. | ||
| Fig. 2 Schematic preparation of Fe3O4/NA and its application for mercury adsorption. | ||
Characterization methods
Inductively coupled plasma atomic emission spectrometry (ICP-AES-6300-Thermo Scientific) was used to determine Hg(II) concentration in aqueous samples. Attenuated Total Reflection FTIR spectra of bare Fe3O4 and Fe3O4/NA were recorded with a Nicolet iS50 FTIR spectrometer (Thermo Scientific, USA) in the range of 4000–800 cm−1, equipped with ZnSe crystal. The spectral resolution was set to 4 cm−1, and 64 scans were collected for each spectrum. The size and morphology of the prepared Fe3O4/NA was studied by field-emission scanning electron microscopy (FESEM, JEOL-7401F) and transmission electron microscopy (TEM, JEM-2200FS). The X-ray diffraction patterns of the products were recorded on a PANalyticalX'Pert diffractometer using Cu Kα radiation with scanning step length of 0.016°. The surface chemical composition of the samples was determined by X-ray photoelectron spectroscopy (XPS) on a VG ESCALAB 250 spectrometer using monochromatic Al Kα X-ray source (1486.8 eV). The Brunauer–Emmett–Teller (BET) specific surface area and pore size distribution were determined by N2 adsorption method using an ASAP 2010 system (Micromeritics Corp., USA) at 77 K. The zeta potential (ξ) and hydrodynamic size distributions were measured with Zetasizer ELSZ-1000. Magnetic behaviour was characterized by Quantum Design PPMS Value Stream Mapping (VSM) system. A 3-Star Orion bench top pH meter (Thermo Scientific) was used for pH measurement during the experiment.Procedure of Hg adsorption
All the adsorption experiments were carried out in duplicate at pH 6 ± 0.1 and 298 K. Briefly, 0.04 g of Fe3O4/NA was added to 30 mL of a 5 mg L−1 Hg(II) solution. The solution was sonicated for a one minute and placed on the rolling mixer (33 rpm) for 1 h. The pH of solution was adjusted by 0.1 M HCl or 0.1 M NaOH and all pH measurements were maintained during the course of experiment. Effect of solution pH and ionic strength was interpreted in the range of (3–9) and (1–100) mM NaCl solutions, respectively. Adsorption kinetics was investigated by varying the equilibration time from 1 to 60 minutes. Adsorption isotherms and thermodynamics were evaluated by varying initial mercury concentration (5–50 mg L−1) and temperature in the range of 298–323 K, respectively.The efficiency of Fe3O4/NA towards combine Hg(II), Cd(II) and As(V) removal was also assessed by spiking 1000 μg L−1 of each Hg(II) Cd(II) and As(V) in groundwater sampled from Yeoncheon, South Korea. This sample contained Ca2+, Na+, Mg2+, K+, Cl−, SO42−, NO3−, and pH was found to be 7.8. After adsorption, the particles were separated in few seconds by applying an external magnetic field and equilibrium concentration of Hg(II) was determined. The interaction between the sorbent (Fe3O4/NA) and Hg(II) is illustrated in Fig. 2.
The adsorption capacity qe (mg g−1), the quantity of Hg(II) adsorbed at equilibrium, was calculated according to following equation:
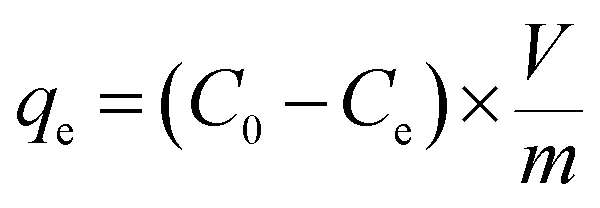 | (1) |
Desorption and regeneration experiments
The desorption experiments were performed with 0.001 M HCl containing 0.3 M thiourea because a thiourea acted as a source of sulphide in reactions with soft metals ions. Briefly, 0.04 g of Hg(II) loaded Fe3O4/NA was washed twice with distilled water to remove the any loosely attached Hg(II) to PET bottle. Then 10 mL of desorbent was added to PET bottle and placed on the rolling mixer (33 rpm) for one hour. Magnet was used to separate Fe3O4/NA in desorbent and Hg(II) in solution was quantified with ICP-AES.Results and discussion
Characterization of Fe3O4/NA
FTIR spectroscopy was utilized for qualitative determination of NA, grafted on the Fe3O4 surface (Fig. S1 (ESI†)). Infrared spectrum describes the symmetrical stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC bands of naphthyridine ring at 1490 and 1450 cm−1, respectively as shown in Fig. 3a. The band at 1253 cm−1 is ascribed to CH deformation.34 Strong band at 1630 cm−1 shows CO stretches of Fe3O4/NA, indicating the carboxylate anion interacting with iron oxide surface as the CO stretches in free carboxylic acid is above 1700 cm−1.35
N and CC bands of naphthyridine ring at 1490 and 1450 cm−1, respectively as shown in Fig. 3a. The band at 1253 cm−1 is ascribed to CH deformation.34 Strong band at 1630 cm−1 shows CO stretches of Fe3O4/NA, indicating the carboxylate anion interacting with iron oxide surface as the CO stretches in free carboxylic acid is above 1700 cm−1.35
 | ||
| Fig. 3 IR spectra (a) and X-ray photoelectron spectroscopy (XPS) (b) of the synthesized Fe3O4/NA. | ||
Fig. 3b represents the wide scan XPS spectra, that reveals the surface composition (w/w), i.e. 17.7% O, 8.7% N and 73.6% C for the NA raw material, whereas 55.03% O, 1.49% N, 29.09% C, and 14.39% Fe for Fe3O4/NA. On the basis of Fe content, it was examined that Fe3O4/NA surface contained 80.11% NA, and the NA in Fe3O4/NA consisted of 68.7% O, 1.85% N, and 36.31% C. The O content in NA coated Fe3O4 was significantly higher as compared to the NA raw material, suggesting that the NA fractions abundant with O based functional groups were selectively coated on the surface of Fe3O4 during the synthesis of Fe3O4/NA. Moreover, by taking into account of high synthesis temperature (80 °C) it was also possible that some fractions of NA degraded to form products with higher contents of O-based functional group which adsorbed preferentially on the surface of Fe3O4.36
The typical SEM and TEM images of Fe3O4/NA are shown in Fig. 4a and b, respectively. The SEM image reveals that the product is composed of irregular shaped rods which are interconnected each other. The TEM image represents the core of Fe3O4 magnetic nanoparticles, having a typical size of ∼10 nm, but the entire Fe3O4/NA particles contain aggregates with no uniform size and fractal feature. The average particle size as determined in suspension by DLS was 1776 nm which was larger than the size measured by TEM. The difference in size was due to both aggregation and water solvation around the particles.
 | ||
| Fig. 4 SEM (a) and TEM (b) images of the prepared Fe3O4/NA. | ||
According to TOC analysis, the actual NA content was found to be ∼0.62 wt% of Fe3O4/NA. The XRD spectrum of bare Fe3O4 was compared with Fe3O4/NA as shown in Fig. 5a. The XRD pattern of the magnetite nanoparticles exactly matched the JCPDS reference no. 19-629. These two patterns were quite similar, and all of the diffraction peaks were indexed as face centered cubic faces of Fe3O4. These results corroborated that NA did not result in phase change in the structure of Fe3O4.
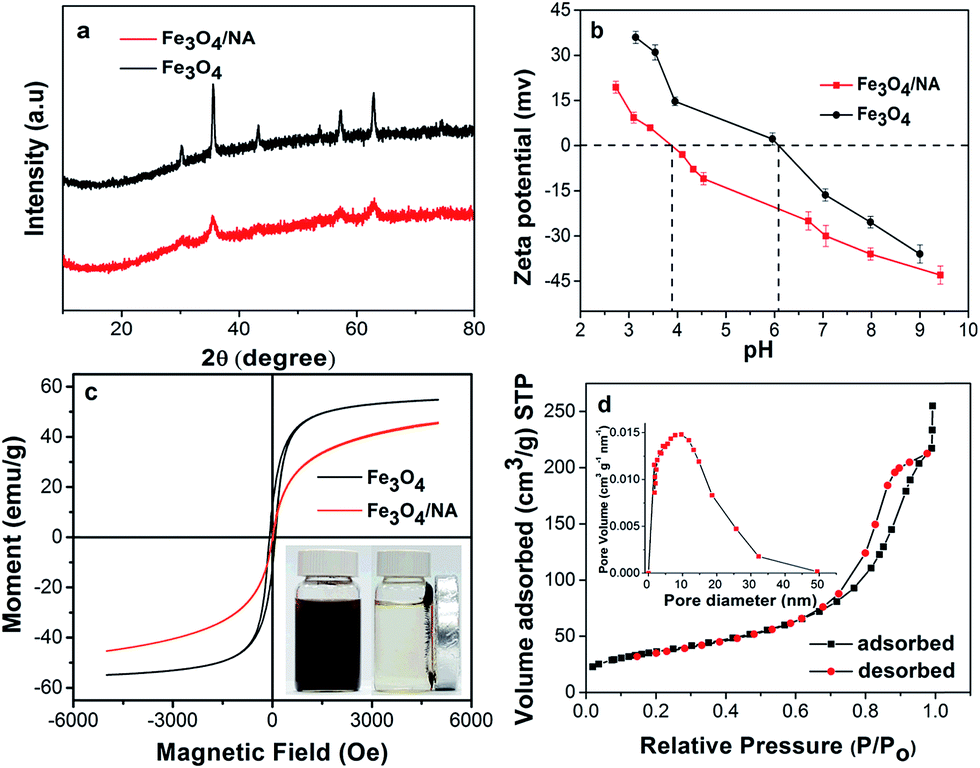 | ||
| Fig. 5 (a) XRD patterns of Fe3O4 and Fe3O4/NA. (b) Zeta potentials of Fe3O4 and Fe3O4/NA as function of pH in 1 mM NaNO3. (c) Hysteresis loop of Fe3O4/NA at 300 K and (inset) dispersed water solution and magnetic separation (d) nitrogen adsorption–desorption isotherm and Barrett–Joyner–Halenda (BJH) pore size distribution plot (inset) of the prepared Fe3O4/NA. | ||
The respective zeta potential of Fe3O4 and Fe3O4/NA under varying pH was measured to investigate the point of zero charge (PZC) as shown in Fig. 5b. The PZC for Fe3O4/NA was found to be around pH 3.9 which was much lower than that of Fe3O4 (pH 6.1). The low PZC value of Fe3O4/NA indicated that the NA cover the surface of Fe3O4 which facilitated more adsorption of positively charged Hg(II) over a wide range of pH values.
To determine the magnetic properties of the sorbent, a VSM was used. The Fe3O4/NA particles were found to be superparamagnetic with a magnetization (M) value of 45 emu g−1 at room temperature (Fig. 5c). The hysteresis loop demonstrated that the coercivity and remanence were almost negligible (Fig. S2 (ESI†)), which is desirable for many practical applications.
The N2 adsorption desorption isotherms and the pore size distribution of Fe3O4/NA are shown in Fig. 5d. The BET analysis revealed that the specific surface area of Fe3O4/NA were to be 126 m2 g−1 which was almost four times higher than pure Fe3O4 (32 m2 g−1). The BET surface area, the large total pore volume (0.035 cm3 g−1) and the pore size (10.84 nm) strongly indicate that the Fe3O4/NA have a mesoporous structure which is desirable for enhanced adsorption performance.
Adsorption kinetics
The adsorption kinetics of Hg(II) to Fe3O4/NA and bare Fe3O4 is presented in Fig. 6a. It could be seen that the adsorption of Hg(II) in two systems increased rapidly in first 10 min of contact time and then maintained the level with subsequent increase in time. Fe3O4/NA achieved the better adsorption efficiencies as compared to bare Fe3O4. Such a fast adsorption could be due to the absence of internal diffusion resistance20 and is favorable for the practical application of Fe3O4/NA as sorbent for removal of toxic metals. The relatively high removal capacity of Fe3O4/NA is expected from their greater surface area which contributes to the increase in the number of binding sites for metal ions. On the basis of adsorption kinetic data, 60 min shaking time was employed in the following experiments to ensure complete equilibrium. In order to investigate the adsorption mechanism the experimental kinetic data of Hg(II) adsorption on bare Fe3O4 and Fe3O4/NA were evaluated by the pseudo-first-order and pseudo-second-order kinetic models.37,38 | ||
| Fig. 6 (a) Effect of time on Hg sorption on bare Fe3O4 and Fe3O4/NA. (b) Pseudo-second order model fitting of sorption kinetic data. | ||
The pseudo first order kinetic model is expressed as follows:
ln(qe − qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − k1t qe − k1t
| (2) |
The pseudo second order kinetic model is expressed as follows:
 | (3) |
The calculated kinetic parameters from both model fittings are listed in Table S1 ESI.† The validity of both models was estimated by comparing the correlation coefficient (R2) values. Here the R2 value for the pseudo-second-order model was higher than that for the pseudo-first-order model. Moreover, the experimentally determined qe,exp (3.63 mg g−1) from the pseudo-second-order model fitting was much closer to the theoretically obtained pseudo-second-order model qe,cal (3.60 mg g−1) as compared to the pseudo first order model qe,exp (0.80 mg g−1). These results predicated that the adsorption system obeyed the pseudo-second-order kinetic model and therefore, supported the assumption that sorption was due to chemisorption instead of mass transport.39,40
Effects of pH and salinity
The effect of pH on the adsorption of Hg(II) onto Fe3O4/NA is shown in Fig. 7a. It was observed that removal capacity was highly dependent on pH. The removal capacity increased with the increasing of pH value and reached a plateau after pH 5. This result suggests that Fe3O4/NA would be useful in contaminated natural waters with broad range of pH values. It is expected that the change in pH of the solution results in forming different Hg aqueous species41 such as Hg2+, HgOH+, and Hg(OH)2 and different surface charges of Fe3O4/NA. When the pH was lowered than the Point of Zero Charge (PZC) of Fe3O4/NA (∼3.9), the surface of Fe3O4/NA was positively charged and had very weak interaction with mercury cations due to protonation of the surface functional groups accompanied with the competition between mercuric ions and H+ or H3O+ ions present in the solution. On the other hand, when pH was higher than the PZC of Fe3O4/NA, the surface of Fe3O4/NA was negatively charged and more functional groups are available for metal cation binding due to deprotonation, resulting in electrostatic attraction between Hg(II) and Fe3O4/NA.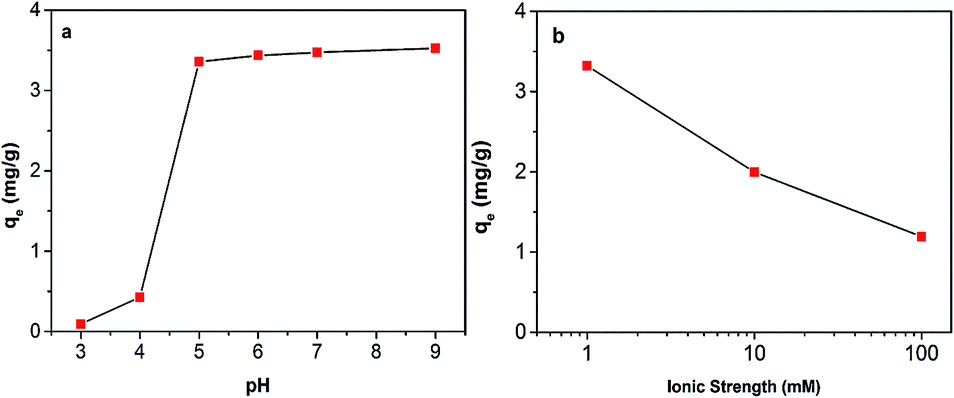 | ||
| Fig. 7 Influence of pH (a) and ionic strength (b) on Hg(II) sorption on Fe3O4/NA. | ||
Since interaction of various chemical compounds and electrolytes may change the surface properties of sorbent materials, removal capacity was investigated using various concentrations of NaCl solutions (Fig. 7b). It was noted that removal of Hg(II) onto Fe3O4/NA gradually decreased as the ionic strength of NaCl solution increased from 1 to 100 mM. The adverse effect of ionic strength is described by two factors: (i) the electrolyte ions (Na+) compete with positively charged mercury ions for the same binding sites and (ii) the ionic strength influences the interfacial potential of mercury metal ions, which would in turn limit their transfer to the Fe3O4/NA surface.42
Effects of initial concentration and adsorption isotherm
The concentration dependence of Hg(II) adsorption onto Fe3O4/NA was studied under the optimized conditions of equilibration time and pH and the results are depicted in Fig. S3a (ESI†). It was observed that the adsorbed amount of Hg(II) on Fe3O4/NA (qe) almost linearly increased with the increasing the initial concentration of Hg(II) and reached a plateau at higher concentrations which might be resulted from saturation of the limited number of binding sites in a fixed amount of adsorbent.39,43 The experimental data relating to the adsorption of Hg(II) onto Fe3O4/NA was interpreted by Langmuir and Freundlich sorption isotherms using equations given below:
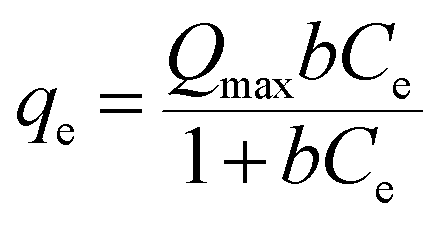 | (4) |
 | (5) |
qe vs. logCe. The corresponding parameters related to the Langmuir and Freundlich adsorption models are given in Table 1. The correlation coefficients (R2) for Langmuir and Freundlich models were 0.98 and 0.94, respectively. The good agreement between the adsorption data and the Langmuir model confirms surface homogeneity, monolayer surface coverage, and no lateral interaction between sorbed molecules.
| Qmax (mg g−1) | Langmuir b (L mg−1) | R2 | KF | Freundlich n | R2 |
|---|---|---|---|---|---|
| 9.52 | 0.50 | 0.98 | 1.90 | 4.97 | 0.94 |
Adsorption thermodynamics
Thermodynamic behavior of the adsorption of Hg(II) onto Fe3O4/NA was evaluated with help of thermodynamic parameters including standard free energy (ΔG°), enthalpy change (ΔH°), and entropy change (ΔS°). These parameters were estimated from the following equations:|
ΔG° = −RTlnKD
| (6) |
 | (7) |
The thermodynamic parameters were given in Table S3 ESI.† The negative values of ΔG° indicate that the adsorption of Hg(II) onto Fe3O4/NA is thermodynamically feasible and spontaneous in nature. The decrease in numerical value of −ΔG° with rise in temperature exhibits that Hg(II) sorption process on Fe3O4/NA is exothermic and less favorable at higher temperatures44 These results correspond to Jeon and coworkers45 who suggested that mercury escape from the surface site increased with rise in temperature and the amount adsorbed decreased, resulting in reduction in boundary layer thickness. The ΔH° parameter was found to be −9.63 kJ mol−1. The negative ΔH° indicates the exothermic nature of the adsorption process of Hg(II) at 25–50 °C. The ΔS° parameter was found to be −15.73 J mol−1 K−1. The negative ΔS° value indicates a decrease in the randomness at the solid/solution interface during the adsorption process of Hg(II) onto Fe3O4/NA.
Material stability and environmental significance
Escape of sorbent fractions into treated water is undesirable. Therefore, in order to ensure the permanent incorporation of NA in Fe3O4/NA matrix, a leakage test was performed under the stated experimental conditions and the contact time was prolonged to 56 hours as shown in Fig. 8a. No leakage of NA was observed, which confirmed the stability of Fe3O4/NA and supporting its use in real environments. | ||
| Fig. 8 Leakage test of Fe3O4/NA (a), the removal efficiency of heavy metals in groundwater (b), (the water contained mixed Hg, Cd and As with initial concentration of 1000 μg L−1 for each metal ion). | ||
The presence of competitive ions in the adsorptive medium may change the environment and solution chemistry of the target metal ions, which affects the adsorption efficiency of conventional adsorbent. Table S4 (ESI†) describes the presence of different ions in groundwater collected from Yeoncheon, South Korea. The performance of the Fe3O4/NA in the real groundwater spiked with mixed heavy metal ions, containing 1000 μg L−1 of each Hg(II), Cd(II) and As(V), was investigated as shown in the Fig. 8b. It was observed that despite the presence of high concentration of various cations and anions in groundwater, not only the Hg(II) but also higher percentage of Cd(II) and As(V) was removed in similar conditions. This suggested that Fe3O4/NA could be used to mitigate other toxic heavy metals in real groundwater systems.
Mechanism of Hg(II) removal
The removal of metal at the adsorbent surface can be attributed to different plausible mechanisms including adsorption, ion exchange, absorption, surface precipitation, coprecipitation, diffusion and etc.46,47 The XPS analysis of Fe3O4/NA-Hg revealed vital information which was valuable in order to investigate the removal mechanism of Hg(II) by using Fe3O4/NA. Fig. 9a shows narrow XPS scan of Hg in 4f region and the peak at 101.5 eV corresponds to binding energy of 4f7/2 of Hg(II),47 indicating Hg(II) is adsorbed onto the Fe3O4/NA. The narrow scan of the binding energy (BE) of oxygen (O1s) of Fe3O4/NA-Hg (Fig. 9b) indicates the relatively stronger intense peak at 530.9 eV in which O atom is present in low binding energy region corresponding to more reduced state.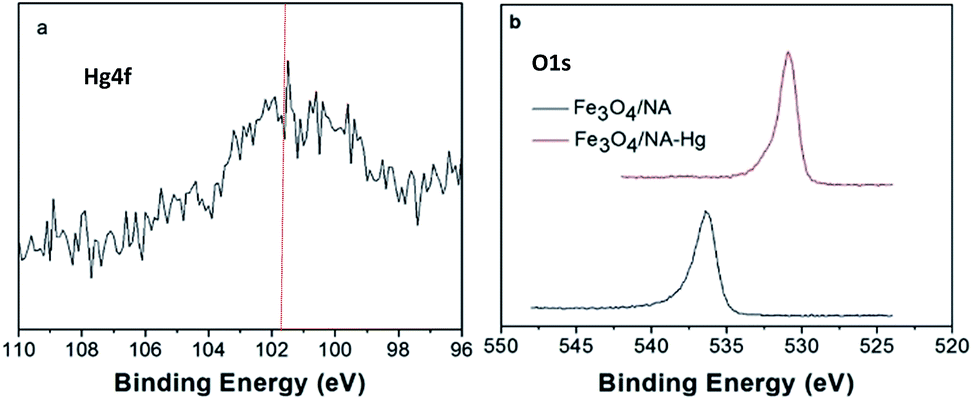 | ||
| Fig. 9 Hg4f narrow scan of Fe3O4/NA-Hg (a), O1s narrow scan before and after adsorption of Hg(II) (b). | ||
This may be due to the formation HgO in which Hg shared electrons with oxygen, which consequently decreases the electronic cloud density of oxygen atom of iron oxide.48 Moreover, the supplemented affinity towards Hg(II) by NA grafted magnetite Fe3O4/NA can also be predicted on the basis of the Hard Soft Acid Base, (HSAB) theory, which classify the Hg(II) as a soft acid due to the large ionic size, high polarizability and low electronegativity, capable to form strong bonds with soft bases containing N and S atoms.49 Therefore, our study on the adsorption of Hg(II) by Fe3O4/NA-Hg showed the participation of oxygen in complexation with heavy metal through XPS analysis with synergistic interaction between N and Hg(II).
Regeneration and reusability
The recycling and reusability is a basic economic necessity for the practical and industrial application of sorbents. In this context, the regeneration and reusability of Fe3O4/NA was verified to assess its application potential in the decontamination of Hg(II) bearing water. Taking into consideration, the negligible adsorption capacity exhibited by Fe3O4/NA at low pH, acid treatment was found to be a feasible approach for regeneration. Therefore, 0.001 M HCl containing 0.3 M thiourea was used as a complexing agent to recover Hg(II) for consecutive five cycles and the desorption efficiency was found to be still above 90%. As shown in Fig. 10a, after first adsorption cycle there was a decrease of ∼6% in removal efficiency, but for the remaining cycles it was remained to ∼93%, representing the excellent regeneration and reusability. The slight reduction in adsorption capacity might be expected due to incomplete desorption of Hg(II) from the surface of Fe3O4/NA. The SEM images of freshly prepared and regenerated samples after fifth cycle are shown in Fig. 10b. After five cycles, the surface of Fe3O4/NA was appeared to be little bit rougher with some broken spheres but it still kept rod-like morphology. Overall, Fe3O4/NA can be easily regenerated, collected in short time and reused several times, which ensures its long-term use for Hg(II) removal from wastewater. | ||
| Fig. 10 Recycling of Fe3O4/NA in the removal of Hg(II) with initial concentration of 1000 μg L−1 (a), SEM images of fresh and recycled Fe3O4/NA (b). | ||
Comparison with other sorbents
In order to verify the effectiveness of Fe3O4/NA as a potential sorbent for Hg(II) Qmax was compared carefully with those of other sorbents reported in previous literature as listed in Table S2 ESI.† It can be seen that the maximum adsorption capacity of Fe3O4/NA is higher than that of clay,50 fly ash,51 rice husk ash,52 activated carbon,53 magnetic nanoparticles modified with 2-mercaptobenzothiazole,54 naphthalimide-functionalized magnetic nanosensor57 and comparable with silica grafted methyl amino ethyl methacrylate56 while lower than dithiocarbamate grafted magnetite particles58 and thiol-modified magnetite beads-porous materials.55 Despite some good benefits of dithiocarbamate and thiol-modified materials, the complex, tedious synthetic procedure and expensive raw materials cost limit their wide scale application.In comparison to others, Fe3O4/NA can be synthesized by the facile and controllable method with economical nalidixic acid and iron precursors at low temperature. In addition, Fe3O4/NA is magnetically retrievable, stable, attain the equilibrium within one hour and can be reused for several times. It is important to mention here that the Fe3O4/NA is capable to reduce the initial Hg(II) (1000 μg L−1) concentration to values lower than 2 μg L−1 in real groundwater. This fact is attributed not only to their adsorption capacity but also because the number of previous studies consider initial mercury concentrations very high which does not reflect exact degree of contamination observed in the actual environment.
Conclusions
For mercury removal from water, we have synthesized nalidixic acid grafted magnetite nanoparticles (Fe3O4/NA) with high surface area by a facile, low cost and time saving route. Fe3O4/NA can be easily separated by an external magnetic field in less than 20 seconds. The Fe3O4/NA nanoparticles showed near complete removal of 1000 ppb of initial Hg(II) and the enhanced rate constants at near neutral pH was due to the increased sorption sites in the presence of nalidixic acid. The adsorption process was found to be exothermic and spontaneous with decreased randomness at the solid-solution interface. The strong affinity towards Hg(II) was attributed to the synergistic effect of the soft interaction (Hg–N) and the formation of HgO. The adsorbed Hg(II) could be effectively desorbed by 0.001 M HCl containing 0.3 M thiourea without the destruction of particles and subsequently be reused for consecutive adsorption–desorption experiments. The Fe3O4/NA can also be used to remediate other heavy metals such as cadmium and arsenic in groundwater. The proposed method has opened a new class of organic reagent based magnetite that can be utilized as a selective complexing agent for various metal ions.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2011-0028723) and “The GAIA Project” by the Korea Ministry of Environment (RE201402059). Additional research funding was supported by basic research support project (4.0010363.01) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology.Notes and references
- P. Holmes, K. A. F. James and L. S. Levy, Sci. Total Environ., 2009, 408, 171–182 CrossRef CAS PubMed.
- G. K. Darbha, A. K. Singh, U. S. Rai, E. Yu, H. Yu and P. C. Ray, J. Am. Chem. Soc., 2008, 130, 8038–8043 CrossRef CAS PubMed.
- F. Zahir, S. J. Rizwi, S. K. Haq and R. H. Khan, Environ. Toxicol. Pharmacol., 2005, 20, 351–360 CrossRef CAS PubMed.
- G. Bayramoǧlu and M. Y. Arica, J. Hazard. Mater., 2007, 144, 449–457 CrossRef PubMed.
- L. I. Sweet and J. T. Zelikoff, J. Toxicol. Environ. Health, Part B, 2001, 4, 161–205 CAS.
- E. Hodgson, A Textbook of Modern Toxicology, Wiley & Sons, 2004 Search PubMed.
- I. Wagner-Döbler, H. Von Canstein, Y. Li, K. N. Timmis and W. D. Deckwer, Environ. Sci. Technol., 2000, 34, 4628–4634 CrossRef.
- USEPA. National primary drinking water regulations. http://www.epa.gov/safewater/contaminants/index.html#inorganic, accessed June 2015.
- M. M. Matlock, B. S. Howerton and D. A. Atwood, J. Hazard. Mater., 2001, 84, 73–82 CrossRef CAS PubMed.
- M. Huebra, M. P. Elizalde and A. Almela, Hydrometallurgy, 2003, 68, 33–42 CrossRef CAS.
- H. Bessbousse, T. Rhlalou, J.-F. Verchère and L. Lebrun, Chem. Eng. J., 2010, 164, 37–48 CrossRef CAS.
- K. Chakrabarty, P. Saha and A. K. Ghoshal, J. Membr. Sci., 2010, 346, 37–44 CrossRef CAS.
- S. Chiarle, M. Ratto and M. Rovatti, Water Res., 2000, 34, 2971–2978 CrossRef CAS.
- P. N. Diagboya, B. I. Olu-Owolabi and K. O. Adebowale, RSC Adv., 2015, 5, 2536–2542 RSC.
- J. Goel, K. Kadirvelu and C. Rajagopal, Environ. Technol., 2004, 25, 141–153 CrossRef CAS PubMed.
- I. Ali, Chem. Rev., 2012, 112, 5073–5091 CrossRef CAS PubMed.
- Y. Wang, J. Ma and K. Chen, Phys. Chem. Chem. Phys., 2013, 15, 19415–19421 RSC.
- S. C. N. Tang and I. M. C. Lo, Water Res., 2013, 47, 2613–2632 CrossRef CAS PubMed.
- D. Maity and D. C. Agrawal, J. Magn. Magn. Mater., 2007, 308, 46–55 CrossRef CAS.
- E. J. Kim, C. S. Lee, Y. Y. Chang and Y. S. Chang, ACS Appl. Mater. Interfaces, 2013, 5, 9628–9634 CAS.
- K. Mandel, F. Hutter, C. Gellermann and G. Sextl, ACS Appl. Mater. Interfaces, 2012, 4, 5633–5642 CAS.
- A. Farrukh, A. Akram, A. Ghaffar, S. Hanif, A. Hamid, H. Duran and B. Yameen, ACS Appl. Mater. Interfaces, 2013, 5, 3784–3793 CAS.
- S. Shin and J. Jang, Chem. Commun., 2007, 4230–4232 RSC.
- S. Shi, Y. Fan and Y. Huang, Ind. Eng. Chem. Res., 2013, 52, 2604–2612 CrossRef CAS.
- J. Zhu, S. Wei, H. Gu, S. B. Rapole, Q. Wang, Z. Luo, N. Haldolaarachchige, D. P. Young and Z. Guo, Environ. Sci. Technol., 2012, 46, 977–985 CrossRef CAS PubMed.
- X. Liu, Q. Hu, Z. Fang, X. Zhang and B. Zhang, Langmuir, 2009, 25, 3–8 CrossRef CAS PubMed.
- L. Zhou, T. L. Thanh, J. Gong, J.-H. Kim, E.-J. Kim and Y.-S. Chang, Chemosphere, 2014, 104, 155–161 CrossRef CAS PubMed.
- S. Sadeghi and E. Sheikhzadeh, Microchim. Acta, 2008, 163, 313–320 CrossRef CAS.
- X. Jia, L. Li and H. Huang, Fenxi Huaxue, 1996, 24, 1218 Search PubMed.
- P. B. Issopoulos, Acta Pharm. Jugosl., 1989, 39, 267–274 CAS.
- S. Shahida, A. Ali, M. H. Khan and M. M. Saeed, Environ. Monit. Assess., 2013, 185, 1613–1626 CrossRef CAS PubMed.
- S. Shahida, A. Ali and M. H. Khan, J. Iran. Chem. Soc., 2013, 10, 461–470 CrossRef CAS.
- A. Ali, Y. Arafat Abbasi, M. Haleem Khan and M. Mufazzal Saeed, Radiochim. Acta, 2010, 98, 513–517 CAS.
- S. Gunasekaran, R. K. Natarajan, R. Rathikha and D. Syamala, Indian J. Pure Appl. Phys., 2005, 43, 503–508 CAS.
- W. Yantasee, C. L. Warner, T. Sangvanich, R. S. Addleman, T. G. Carter, R. J. Wiacek, G. E. Fryxell, C. Timchalk and M. G. Warner, Environ. Sci. Technol., 2007, 41, 5114 CrossRef CAS PubMed.
- E. Illés and E. Tombácz, Colloids Surf., A, 2003, 230, 99–109 CrossRef.
- Y. S. Ho and G. McKay, Water Res., 1999, 33, 578–584 CrossRef CAS.
- Y. S. Ho and G. McKay, Process Biochem., 1999, 34, 451–465 CrossRef CAS.
- S. Pan, H. Shen, Q. Xu, J. Luo and M. Hu, J. Colloid Interface Sci., 2012, 365, 204–212 CrossRef CAS PubMed.
- Y. S. Ho, J. Hazard. Mater., 2006, 136, 681–689 CrossRef CAS PubMed.
- F. F. S. Zhang, J. O. Nriagu and H. Itoh, Water Res., 2005, 39, 389–395 CrossRef CAS PubMed.
- Z. Reddad, C. Gerente, Y. Andres and P. Le Cloirec, Environ. Sci. Technol., 2002, 36, 2067–2073 CrossRef CAS PubMed.
- B. S. Inbaraj and N. Sulochana, J. Hazard. Mater., 2006, 133, 283–290 CrossRef CAS PubMed.
- A. Sari and M. Tuzen, J. Hazard. Mater., 2009, 171, 500–507 CrossRef CAS PubMed.
- C. Jeon and H. P. Kwang, Water Res., 2005, 39, 3938–3944 CrossRef CAS PubMed.
- C. S. Kim, J. J. Rytuba and G. E. Brown Jr, J. Colloid Interface Sci., 2004, 271, 1–15 CrossRef CAS PubMed.
- Y. Kim and Y. J. Lee, J. Colloid Interface Sci., 2014, 430, 193–199 CrossRef CAS PubMed.
- C. R. Collins, D. M. Sherman and K. V. Ragnarsdottir, J. Colloid Interface Sci., 1999, 219, 345–350 CrossRef CAS PubMed.
- P. Miretzky and A. F. Cirelli, J. Hazard. Mater., 2009, 167, 10–23 CrossRef CAS PubMed.
- W. U. Senevirathna, H. Zhang and B. Gu, Water, Air, Soil Pollut., 2011, 215, 573–584 CrossRef CAS.
- A. K. Sen and A. K. De, Water Res., 1987, 21, 885–888 CrossRef CAS.
- Q. Feng, Q. Lin, F. Gong, S. Sugita and M. Shoya, J. Colloid Interface Sci., 2004, 278, 1–8 CrossRef CAS PubMed.
- X. Lu, J. Jiang, K. Sun, J. Wang and Y. Zhang, Mar. Pollut. Bull., 2014, 78, 69–76 CrossRef CAS PubMed.
- H. Parham, B. Zargar and R. Shiralipour, J. Hazard. Mater., 2012, 205–206, 94–100 CrossRef CAS PubMed.
- J. Dong, Z. Xu and F. Wang, Appl. Surf. Sci., 2008, 254, 3522–3530 CrossRef CAS.
- L. Zhao, J. Sun, Y. Zhao, L. Xu and M. Zhai, Chem. Eng. J., 2011, 170, 162–169 CrossRef CAS.
- J. Zhao, B. Zhu, H. Yu, L. Yan, Q. Wei and B. Du, J. Colloid Interface Sci., 2013, 389, 46–52 CrossRef CAS PubMed.
- P. Figueira, C. B. Lopes, A. L. Daniel-da-Silva, E. Pereira, A. C. Duarte and T. Trindade, Water Res., 2011, 45, 5773–5784 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra25927d |
| This journal is © The Royal Society of Chemistry 2016 |