DOI:
10.1039/C5RA25909F
(Paper)
RSC Adv., 2016,
6, 8495-8502
Synthesis of bis(propargyl) aromatic esters and ethers: a potential replacement for isocyanate based curators†
Received
4th December 2015
, Accepted 23rd December 2015
First published on 5th January 2016
Abstract
This study reports the synthesis and characterization of a novel class of non-isocyanate curing agents based on bis-propargyl aromatic esters 2a–e and ethers 4a–c. A total of eight non-isocyanate curators were prepared from the reaction of respective dicarboxy or dihydroxybenzene with propargyl bromide in the presence of potassium carbonate with good yields. The structure and purity of the synthesized compounds and the corresponding intermediates were confirmed by spectral (IR and NMR), thermal (DSC) and chromatographic techniques (HPLC & GC-MS). Furthermore, kinetics of the curing reaction between glycidyl azide polymer (GAP) and the synthesized alkynes (4b, 4c) were studied using time-resolved FT-IR spectroscopy as a function of time at 303, 323 and 333 K. It was found that the curing reaction was faster when the temperature was increased. Kinetic parameters of the curing reaction, such as the reaction order and activation energy, were calculated for the GAP–4a and GAP–4c systems. All the curing reactions followed first order kinetics and the corresponding activation energy of the curing reaction for the systems was found to be 15.56 and 13.22 kcal mol−1. For comparison, curing studies were performed for GAP with a conventional curator Desmodur N-100. GAP cured with non-isocyanate curators offered good mechanical properties compared to GAP cured with isocyanate (N-100). The advantage of these new curing systems is that they do not require catalyst and there is no need for specific environmental conditions. Based on these studies, 1,4-bis(2-propynyloxy)benzene (4b) has the most potential as a non-isocyanate curator for azide polymeric binders.
1 Introduction
There is constant effort towards developing new high performance composite propellant formulations to meet future requirements. The performance of composite propellant formulations can be enhanced by adding energetic oxidizers such as ammonium dinitramide (ADN), hydrazinium nitroformate (HNF) and an energetic binder such as GAP. GAP is usually cured by reaction with polyisocyanates to form a polyurethane network. However, isocyanates are hazardous and moisture sensitive. They react with moisture to release CO2 and form voids in the cured propellant, leading to poor mechanical properties. For these reasons, use of non-isocyanate curing agents are of great interest.1–3 In particular, for ammonium dinitramide (ADN), such non-isocyanate curing agents would be very useful due to the severe compatibility issues between ADN and isocyanates. Therefore, an alternate approach was perceived to exploit the 1,3-dipolar cycloaddition reaction between the azide group of GAP and triple bond of alkynes (Huisgen reaction) forming 1,2,3-triazoles. This is a versatile tool in polymer chemistry for realizing cross-linked networks without any side reaction and is a prime example of click chemistry.4–6
Poly-functional alkynes are potential isocyanate free curing agents for hydroxyl functional azido polymers, which are cured by thermal, non-catalyst reaction with alkynes to form triazole cross-linked energetic binders. Azido polymers can be cross-linked through some or all the azido groups with multifunctional dipolarophiles having reactive groups selected from acrylic and acetylenic esters or amides to produce a polymer material comprised triazoline or triazole groups. Triazole formation is favored by electron donating groups on the azide, whereas electron withdrawing groups are favored on alkynes.7,8 The presence of electron withdrawing groups at the vicinity of alkynes is much preferred due to its better reactivity towards cycloaddition reactions at ambient temperature. In addition to this, introduction of nitro groups in the acetylenic moiety adds more energy to propellant systems. The presence of triazole groups in the cross-linked polymer is expected to enhance the thermal stability and burn rate.9 Earlier studies have reported10,11 that the curing GAP with alkynes, such as bis-propargylsuccinate (BPS), 1,4-bis(1-hydroxypropargyl)benzene (BHPB) and bisphenol-bis(propargyl ether) (BABE), to form a triazole network with azide polymers. ADN/GAP propellant cured with BPS was investigated and found that the burning rate was slightly higher than isocyanate cured propellants.3 Potinus Heike12 et al. studied heat generation rates of GAP with either isocyanate or BPS by heat flow microcalorimetry and their compatibility with ADN. This study reports the synthesis of alkyne based bis-propargyl aromatic esters 2a–e and ethers 4a–c with the aim of creating an alternative to existing isocyanate curing agents for energetic azido polymers. Furthermore, we also report kinetics for the curing reaction between GAP and selected curators (4b, 4c) and compared these results with the curing reaction between GAP and Desmodur N-100 using quantitative FTIR spectroscopy.
2 Results and discussion
Synthesis of compounds 2a–e and 4a–c
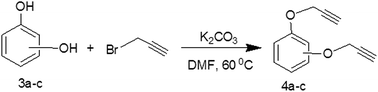
The study presented herein involves synthesis and characterization of bis-propargyl aromatic esters 2a–e and ethers 4a–c, which are potential non-isocyanate curators for energetic azido binders for development in advanced smokeless composite rocket propellant systems. The compounds 2a–e and 4a–c were synthesized by reacting corresponding dicarboxy or dihydroxybenzene with propargyl bromide under mild reaction conditions. The products 2b, 2c and 2e were synthesized for the first time and characterized (Scheme 1). The compounds 2a and 2d were synthesized by modifying literature methods.13,14 The reported methods involve the use of expensive 4-(dimethylamino)pyridine (DMAP) as base following a tedious workup process. Our method used K2CO3 as the base and a simple workup process that obtained good yields (Tables 1 and 2). The compound 4c was synthesized (Scheme 2) for the first time along with compounds 4a and 4b based on reported methods.15,16 The synthesis route and its structural characterization are presented in this study. For comparison, intermediate compounds 1b, 1c, 1d and 3c were also synthesized based on literature procedures.7,17–20
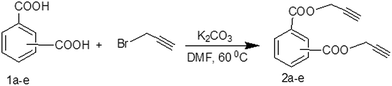 |
| | Scheme 1 Synthesis of bis-propargyl-aromatic esters (2a–e). | |
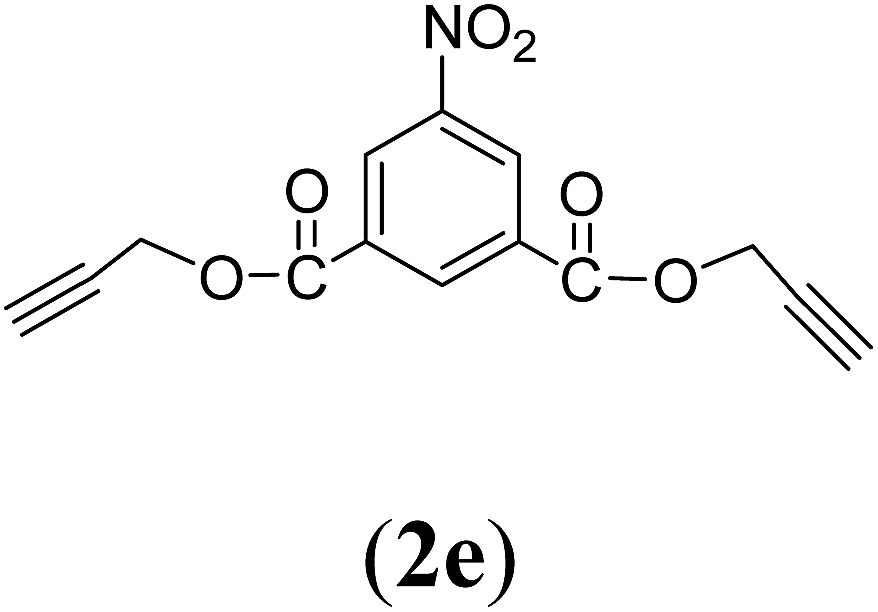
Table 1 Structures and yield of compounds 2a–e
| Entry |
–Ar– |
Product |
m.p.a (°C) |
Yieldb (%) |
| m.p. was determined using the DSC technique. Yields are based on column purification. |
| 1 |
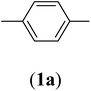 |
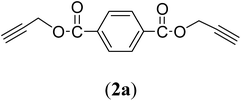 |
110.5 |
54 |
| 2 |
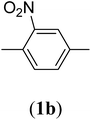 |
 |
88.38 |
61 |
| 3 |
 |
 |
89.98 |
62 |
| 4 |
 |
 |
85.53 |
85 |
| 5 |
 |
 |
145.54 |
70 |
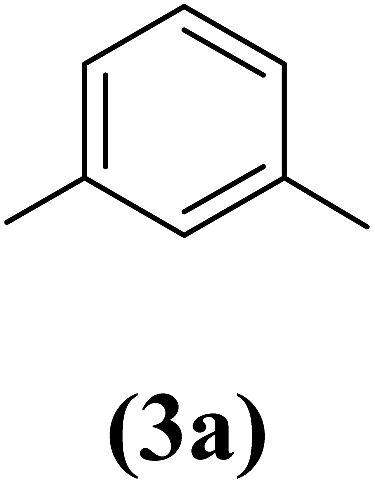
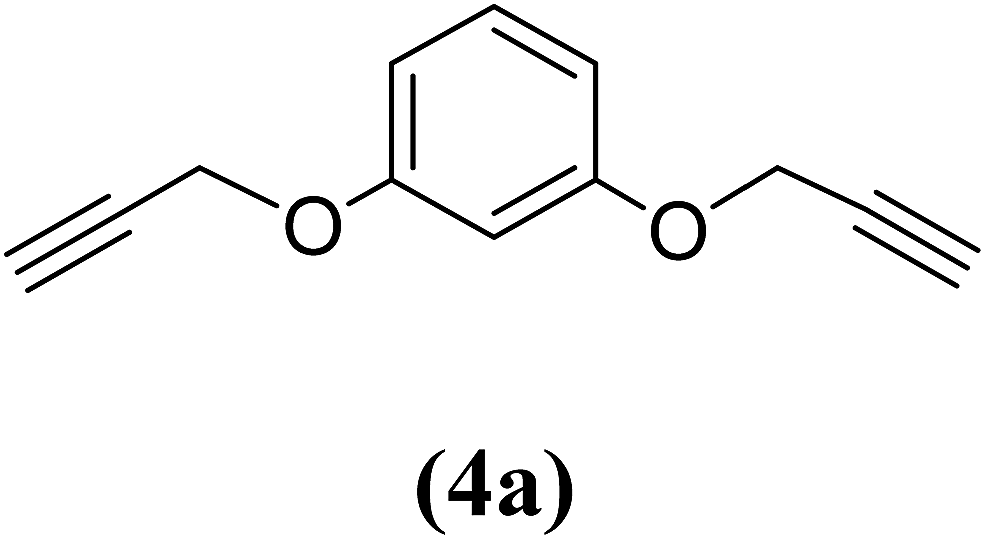
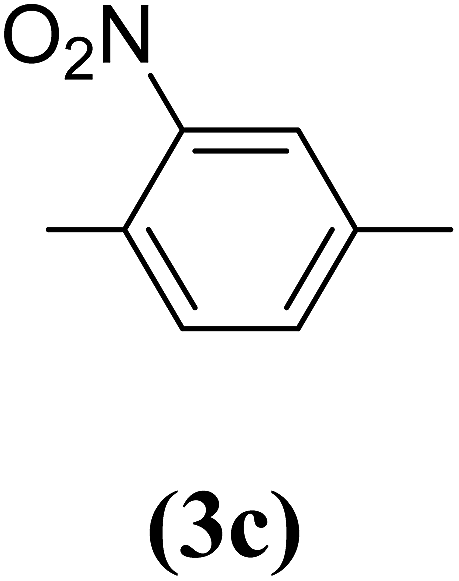
Table 2 Structures and yield of compounds 4a–c
| Entry |
–Ar– |
Product |
m.p.a (°C) |
Yieldb (%) |
| m.p. was determined using the DSC technique. Yields were based on column purification. |
| 1 |
 |
 |
40.69 |
47 |
| 2 |
 |
 |
48.92 |
56 |
| 3 |
 |
 |
76.31 |
46 |
 |
| | Scheme 2 Synthesis of bis-propargyl-aromatic ethers (4a–c). | |
Structural characterization
The structure of compounds 2a–e and 4a–c was confirmed by IR, NMR and mass spectroscopy. FTIR spectra of compounds 2a–e show presence of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– bond with absorption in the range 2127–2133 cm−1, whereas alkyne proton (–CC–H) stretching absorption is in the range of 3269–3294 cm−1. The ketonic C
C– bond with absorption in the range 2127–2133 cm−1, whereas alkyne proton (–CC–H) stretching absorption is in the range of 3269–3294 cm−1. The ketonic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the synthesized nitro dicarboxylic acids (intermediates) were at 1705 cm−1, whereas the CO group of the esters exhibited stretching absorption in the frequency range of 1716–1785 cm−1. FTIR results showed the bis-propargyl aromatic ethers 4a–c showed characteristic absorption peak of –CCH stretching between 3282 and 3289 cm−1, –CC– bond absorption peak at 2127–2133 cm−1 and the C–O–C absorption peak between 1030 and 1091 cm−1. Compounds 2b, 2c, 2e & 4c showed peaks at 1546 and 1328 cm−1 corresponding to NO2 stretching frequencies. All protons were observed in the 1H-NMR spectra of compounds 2a–e and 4a–c with the expected chemical shifts and integral values. The 13C-NMR spectra of compounds 2a–e and 4a–c had a quantity of signals that was consistent with the number of carbon atoms in the molecule (ESI†). The products 2a–e and 4a–c were subjected to GC-MS analysis. The results obtained from the gas chromatograph showed a single peak and the corresponding mass spectra of compounds 2a–e and 4a–c showed a molecular ion peak M+ at m/z in agreement with their molecular formula. Purity of compounds 2a–e and 4a–c was determined by RP-HPLC and was found to be greater than 98%.
O group of the synthesized nitro dicarboxylic acids (intermediates) were at 1705 cm−1, whereas the CO group of the esters exhibited stretching absorption in the frequency range of 1716–1785 cm−1. FTIR results showed the bis-propargyl aromatic ethers 4a–c showed characteristic absorption peak of –CCH stretching between 3282 and 3289 cm−1, –CC– bond absorption peak at 2127–2133 cm−1 and the C–O–C absorption peak between 1030 and 1091 cm−1. Compounds 2b, 2c, 2e & 4c showed peaks at 1546 and 1328 cm−1 corresponding to NO2 stretching frequencies. All protons were observed in the 1H-NMR spectra of compounds 2a–e and 4a–c with the expected chemical shifts and integral values. The 13C-NMR spectra of compounds 2a–e and 4a–c had a quantity of signals that was consistent with the number of carbon atoms in the molecule (ESI†). The products 2a–e and 4a–c were subjected to GC-MS analysis. The results obtained from the gas chromatograph showed a single peak and the corresponding mass spectra of compounds 2a–e and 4a–c showed a molecular ion peak M+ at m/z in agreement with their molecular formula. Purity of compounds 2a–e and 4a–c was determined by RP-HPLC and was found to be greater than 98%.
Curing kinetics of GAP with bis-propargyl ethers
Triazole cross-linked networks are readily formed by the 1,3-dipolar (3 + 2) cycloaddition between the azide group of GAP and triple bond of alkynes without a catalyst. The possible mechanism of formation of the triazole ring is shown in Scheme 3. The required reaction time for formation of triazole cross-linked networks was from 24 h to one week. This is due to the low reactivity of acetylenic compounds towards 1,3-dipolar cycloaddition reactions. The acetylene bond was deactivated by introducing electron withdrawing groups in the vicinity of the molecule.8 Therefore, deactivating groups, such as nitro, carbonyl and aryl moiety, are intentionally introduced to meet the required curing conditions in the temperature range of 35–50 °C.21
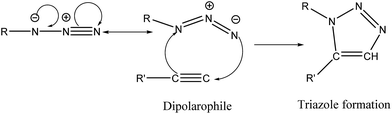 |
| | Scheme 3 Possible mechanism of curing reaction between GAP and an alkyne compound. | |
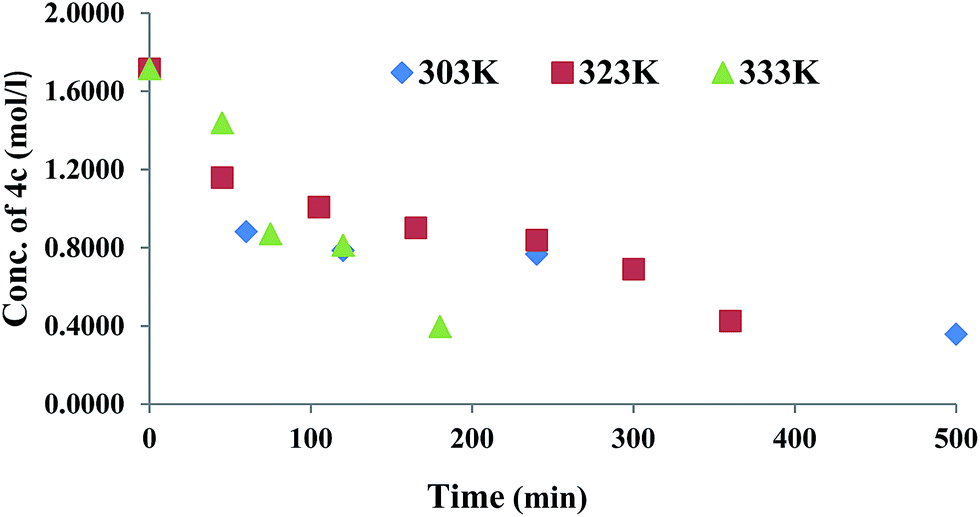
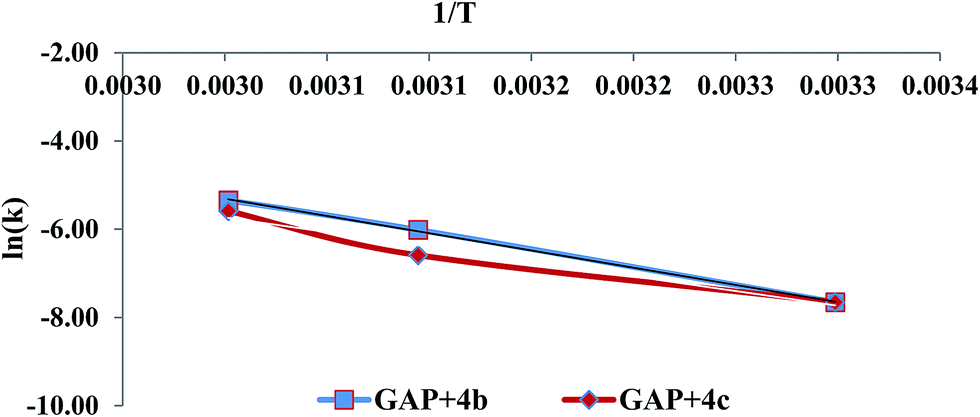
GAP (molecular weight ∼ 1800) and curator 4b or 4c (30% of GAP, w/w) were mixed at room temperature and degassed by applying vacuum. The curing reaction was monitored by FTIR spectroscopy as a function of time at different temperatures (303, 323 and 333 K). The curing reaction was monitored by observing the change in the intensity of the absorption band at 3285 cm−1 that corresponds to the alkyne proton (–CCH) and found to be decreased as the curing reaction proceeded (Fig. 1). The absorption peak at 2094 cm−1 indicates the presence of residual azido groups in the GAP. Characteristic peaks of the triazole ring after curing were observed in the range of 1500–1300 cm−1 corresponding to NN and CC bonds. Curing kinetics of triazole formation between GAP and curator was carried out by evaluating the change in the intensity of alkyne proton (–CCH) stretching band at 3285 cm−1. Intensity of the absorption band for –CCH stretching was calculated by taking the integral of the band between 3310 and 3100 cm−1 using OMNIC software.22 The standard equation was used to analyze the curing profile of GAP with curators 4b or 4c.23,24 A plot of change in concentration of 4b and 4c as a function of time at different temperatures is shown in the Fig. 2 and 3. This indicates that at room temperature (303 K), the curing reaction required 18–30 h for gel formation. As the cure temperature increased, the curing time decreased to 3 h. It was observed that gelation occurred after conversion of 55–60% curing and this value can be served as a semi-quantitative estimate of the gel point. The required curing time for gel formation at different temperatures for the GAP–4b and GAP–4c systems are summarized in Table 3 along with conventional curators for comparison. The results indicate that gel time was faster for nitro substituted curator (4c) than that of without the nitro group (4b). The rate of reaction of curator with and without an electron deficient group (NO2) was studied (Table 4). For example, the curator having a nitro group (4c) showed a higher rate of reaction as compared to a curator without a nitro group (4b). The rate constant at different temperatures is given by the Arrhenius equation, ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k = lnA − (Ea/RT). The activation energy (Ea) for the curing reaction was calculated from the Arrhenius equation by plotting ln(k) versus 1/T (Fig. 4). The measured activation energy for the curing reaction of the GAP–4b and GAP–4c systems was 15.56 and 13.22 kcal mol−1, respectively. This is further evidence of the effect of the nitro group present in the curators.
k = lnA − (Ea/RT). The activation energy (Ea) for the curing reaction was calculated from the Arrhenius equation by plotting ln(k) versus 1/T (Fig. 4). The measured activation energy for the curing reaction of the GAP–4b and GAP–4c systems was 15.56 and 13.22 kcal mol−1, respectively. This is further evidence of the effect of the nitro group present in the curators.
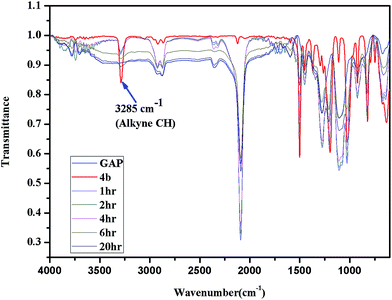 |
| | Fig. 1 FTIR spectrum of pure GAP and GAP cured with 4b at different time intervals. | |
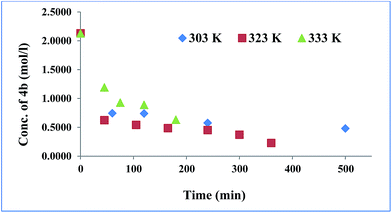 |
| | Fig. 2 Change in concentration of 4b during GAP curing. | |
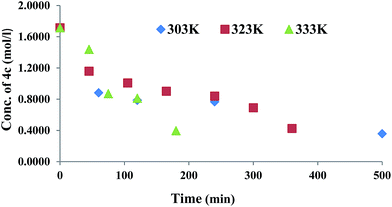 |
| | Fig. 3 Change in concentration of 4c during GAP curing. | |
Table 3 Gel time profile for curing of GAP-N100 and GAP–4b/4c
| Temperature (K) |
GAP + 4b |
GAP + 4c |
GAP + N-100 + catalyst |
| 303 |
23 h |
25 h |
30 h |
| 323 |
3.5 h |
3.0 h |
— |
| 333 |
2.5 h |
2.3 h |
3.7 h (ref. 25) |
Table 4 The rate constants for the GAP–4b & GAP–4c systems
| Temperature (K) |
GAP + 4b system s−1 |
GAP + 4c system s−1 |
| 303.15 |
0.0005 |
0.00047 |
| 323.15 |
0.0024 |
0.00138 |
| 333.15 |
0.0048 |
0.00375 |
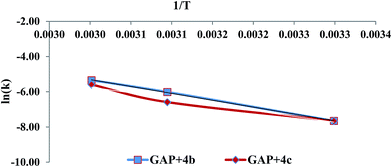 |
| | Fig. 4 Plot of ln(k) verses 1/T for the two systems. | |
Fig. 5 shows the GAP cured with isocyanate in the presence of catalyst at room temperature without humidity control. Similarly, GAP cured with non-isocyanate (4b) curator under the same condition without catalyst for comparison. It was observed that GAP cured with 4b in absence of catalyst was very smooth and clean, whereas isocyanate based curator with GAP showed voids and evacuation of gases.
 |
| | Fig. 5 GAP cured with isocyanate and non-isocyanate under the same experimental conditions. | |
Furthermore, experiments were carried out to observe the difference between curing of GAP with conventional curator and non-isocyanate curator. GAP was cured with isocyanate (N-100) in the presence of catalyst dibutyltindilaurate (DBTDL) at room temperature under moisture-free environmental conditions. Similarly, GAP was cured with a non-isocyanate curator at room temperature without moisture control. The results of thermal and mechanical property analysis of cured samples are presented in Table 5. GAP cured with non-isocyanate curator revealed good tensile and E-mod mechanical properties compared to GAP cured with isocyanate (N-100). However, the percentage of elongation decreased, which could be optimized with varying the amount of curator. The GAP-aromatic bis(propargyl) ethers curing procedure appeared to be simple and could be an alternate to conventional isocyanate based curing systems. In addition to this, these new non-isocyanate based curing systems do not require catalyst and specific humidity control and also the curing reaction occurs at a relatively low temperature (30–50 °C).
Table 5 Thermal and mechanical properties of GAP cured with isocyanate and non-isocyanate curing agents
| Cured polymer samplesa |
DSC exothermic energy release (J g−1) |
Tmax (°C) |
E-mod (kgf cm−2) |
Tensile (kgf cm−2) |
Max. force (kgf) |
Elongation at max. (%) |
Elongation (%) |
| The mixture kept at room temperature for 2 days and post curing for 3 days at 50 °C. |
| GAP + N-100 |
2424 |
250 |
2.14 |
2.52 |
0.35 |
146.61 |
148.47 |
| GAP + 4b |
1499 |
258 |
83.8 |
36.49 |
9.52 |
47.60 |
47.78 |
| GAP + 4c |
2260 |
257 |
58.3 |
18.66 |
5.18 |
35.71 |
35.84 |
3 Conclusions
Bis-propargyl aromatic esters 2a–e and ethers 4a–c were successfully synthesized and fully characterized by FT-IR, NMR, MS and elemental analysis. Reverse phase HPLC analysis of the synthesized compounds showed purity >98%. GC-MS analysis of compounds 2a–e and 4a–c showed a molecular ion peak M+ at m/z in agreement with their molecular formula. The effects of temperature on the curing reaction were studied independently for GAP-N100 and GAP–4b. The results indicated that at room temperature (303 K), 18–30 h were required for gel formation (gel point). It is clearly shown that the gelling time of the newly synthesized curator based binder system decreased as temperature was increased.
The influence of electron deficient groups present in the curators was studied. The results indicated that curators having a nitro group (4c) showed a higher rate of reaction as compared curators without a nitro group (4b). Furthermore, the activation energy for the curing reaction of GAP–4b and GAP–4c systems were 15.56 and 13.22 kcal mol−1, respectively, which was further evidence for an enhanced effect of a nitro group present in the curator. It has been observed that GAP cured with 4b in the absence of catalyst is very smooth and clean, whereas isocyanate based cured GAP showed voids and evacuation of gases.
The results of thermal and mechanical property analysis of GAP cured with N100 or non-isocyanate curators revealed good mechanical properties as compared to conventional curing systems. It has been demonstrated that the GAP-aromatic bis(propargyl) ethers curing procedure appears to be simple and could be an alternative to conventional isocyanate based curing systems. In addition to this, these new non-isocyanate based curing systems do not require any catalyst and specific humid control and also the curing reaction occurred at a relatively low temperature (30–50 °C).
4 Experimental
Materials and methods
Raw materials such as propargyl bromide (80% solution in toluene), potassium carbonate (anhydrous), resorcinol, hydroquinone, p-toluic acid, terephthalic acid, isophthalic acid were purchased from M/s Merck. GAP–diol (molecular weight ∼ 1800) was synthesized in our laboratory. All reagents and chemicals used in the present study were AR grade.
FT-IR spectra were obtained on a Nicolet FTIR-5700 spectrophotometer using a potassium bromide (KBr) matrix/ATR. 1H and 13C NMR spectra were obtained using a Varian 300 MHz instrument in deuterated chloroform/dimethylsulfoxide (DMSO) wherein tetramethylsilane (TMS) was used as the internal standard. Differential Scanning Calorimetery (DSC) was recorded on a DSC-7 Perkin Elmer instrument at a heating rate of 10 °C min−1 in a nitrogen atmosphere. Elemental analysis was recorded using an EA CHNS Model Vario Micro Cube. Gas chromatography-mass spectroscopy (GC-MS) studies were carried out on a Perkin Elmer, Clarus 500 instrument operating at 200 °C source temperature and helium as the carrier gas. High performance liquid chromatography (HPLC) studies were undertaken on an Ultimate_3000 Dionex HPLC system, at 25 °C operating temperature using a reverse phase C-18 column (4 mm × 250 mm), mobile phase acetonitrile/water (60:40), flow rate 1 mL per minute, injection volume 10 μL in isocratic mode. GAP–propargyl ester/ether (30% of GAP, w/w) without catalyst were mixed at room temperature. Curing of GAP with Desmodur N-100 was also carried out at room temperature in the presence of catalyst dibutyltin dilaurate (DBTDL). FTIR spectra were obtained at different time intervals for both the samples and the intensity of the absorption band were calculated using OMNIC software. Mechanical properties, viz., tensile strength and percentage elongation of the cured polymers were evaluated using a Universal Testing Machine (Hounsfield, H25KS).
General procedure for synthesis of bis(prop-2-ynyl)benzene dicarboxylate (2a–e)
Aromatic dicarboxylic acid (1a–e, 0.06 mol) was dissolved in 100 mL of dimethyl formamide. To this solution, K2CO3 (0.14 mol) was added and stirred at 60 °C for 30 min. Then, propargyl bromide (0.15, mol) was added dropwise and the reaction mixture was heated continuously at 60 °C for 7–10 h. The reaction progress was monitored by TLC. After the reaction was completed, the reaction mixture was cooled to room temperature and filtered. The filtrate was quenched with crushed ice and the solid separated was filtered, washed with cold water and dried. The crude products were purified by column chromatography using n-hexane/ethyl acetate as the eluent.
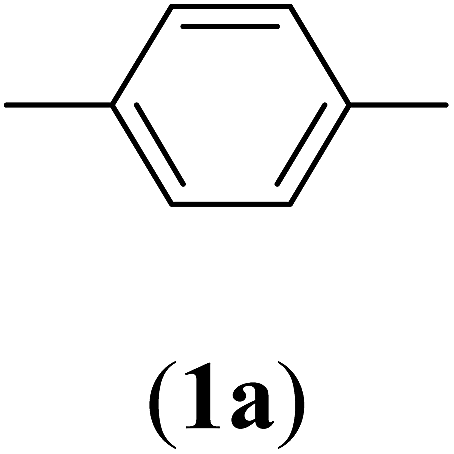
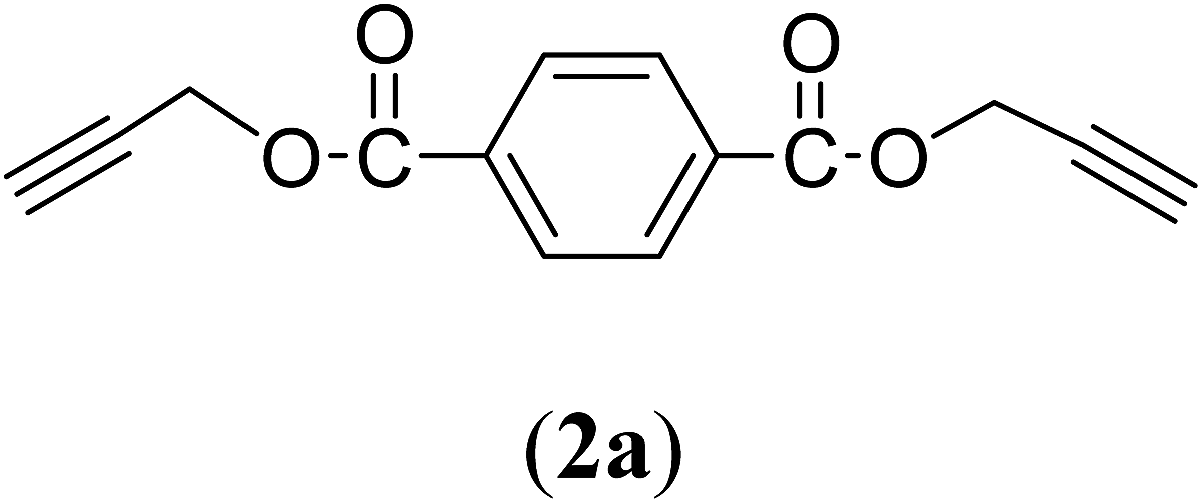
Bis(prop-2-ynyl)benzene-1,4-dicarboxylate (2a). 15.6 g, 54% yield; white solid; m.p. 110.5 °C; Rf: 0.72 (20% EtOAc/n-hexane); 1H-NMR (300 MHz, CDCl3) δ: 2.55–2.56 (t, 2H, CH), 4.96 (d, 4H, CH2), 8.15 (s, 4H, aromatic) ppm; 13C-NMR (300 MHz, CDCl3), δ: 52.83 (CH2), 75.36, 77.31 (CH), 129.84, 133.51 (aromatic), 164.93 (CO) ppm; IR (KBr): 3250, 3120, 2937, 2870, 2127, 1716, 1267, 653 cm−1; GC-MS (EI, 70 eV) m/z: 242 [M+]; elemental anal. calcd (%) for C14H10O4: C, 69.42; H, 4.13; found: C, 69.14; H, 4.08.
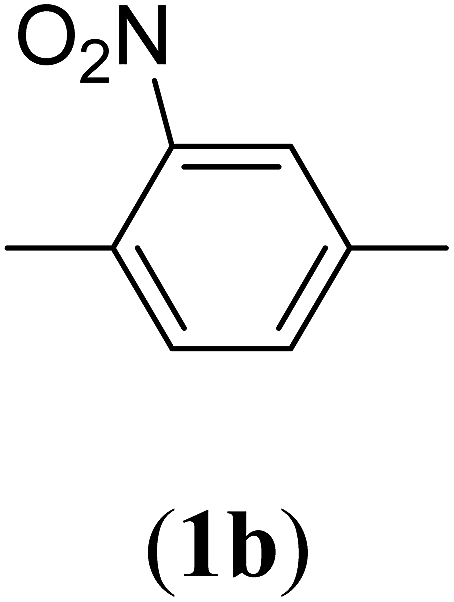
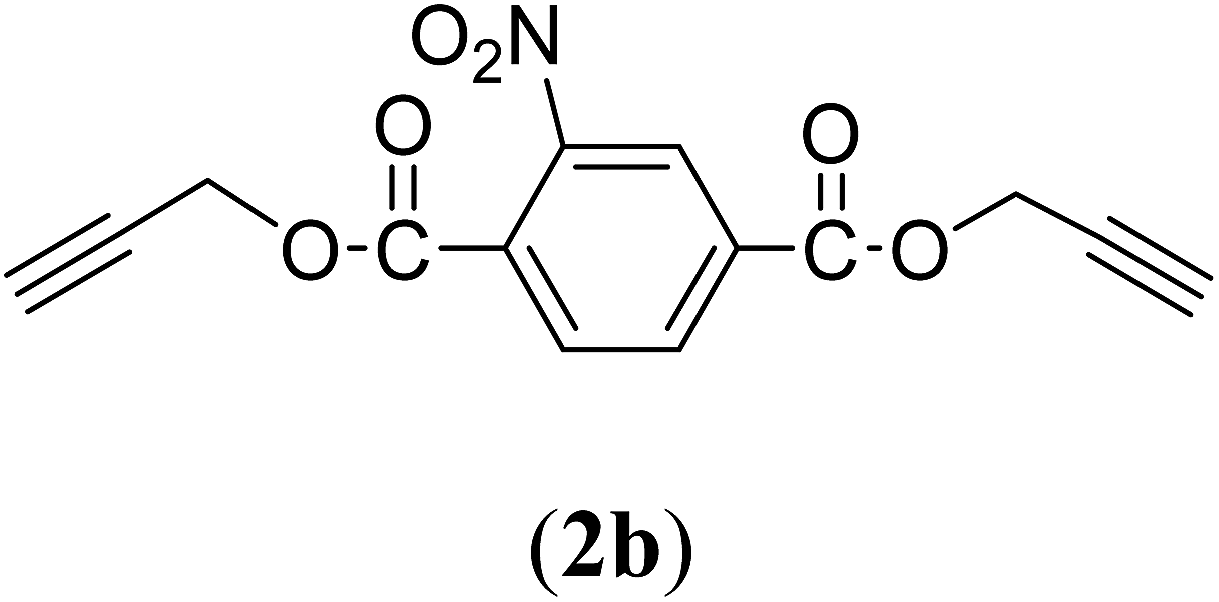
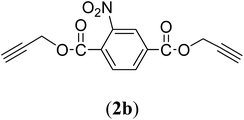
Bis(prop-2-ynyl)-2-nitrobenzene-1,4-dicarboxylate (2b). 1.67 g, 61% yield; white solid; m.p. 88.38 °C; Rf: 0.61 (20% EtOAc/n-hexane); 1H-NMR (300 MHz, CDCl3) δ: 2.60–2.61 (t, 2H, CH), 4.95–4.99 (dd, J = 10.40, 2.47 Hz, 4H, CH2), 7.82–7.85 (d, 1H, Ar), 8.35–8.38 (dd, J = 7.96, 1.64 Hz, 1H, Ar), 8.62 (s, 1H, Ar) ppm; 13C-NMR (300 MHz, CDCl3) δ: 52.77 (CH2), 75.30, 77.38 (CH), 128.79, 129.97, 131.11, 134.37 (aromatic), 164.84 (CO) ppm; IR (KBr) ν: 3279, 3117, 3059, 2939, 2883, 2133, 1732, 1616, 1539, 1363, 1226, 1062, 655 cm−1; GC-MS (EI, 70 eV) m/z: 287 [M+]; elemental anal. calcd (%) for C14H9NO6: C, 58.53; H, 3.13; N, 4.87; found: C, 58.18; H, 2.95; N: 4.86.
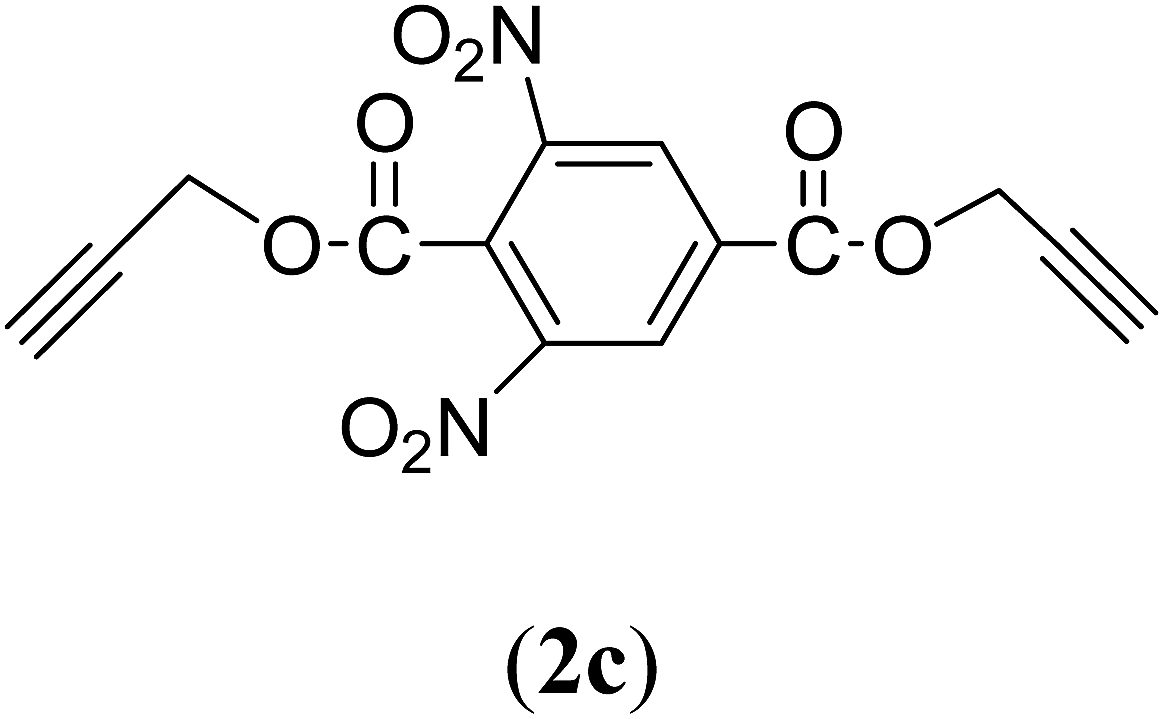
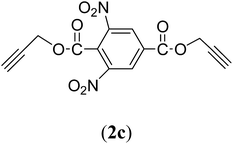
Bis(prop-2-ynyl)-3,5-dinitrobenzene-1,4-dicarboxylate (2c). 1.62 g, 62% yield; cream solid; m.p. 89.98 °C; Rf: 0.9 (20% EtOAc/n-hexane); 1H-NMR (300 MHz, CDCl3) δ: 2.62–2.63 (t, J = 2.48 Hz, 2H, CH), 5.05–5.06 (d, 4H, CH2), 9.23–9.24 (t, 2H, Ar) ppm; 13C-NMR (300 MHz, CDCl3) δ: 54.04 (CH2), 76.37, 77.25 (CH), 122.70, 129.56, 132.99, 148.65 (aromatic), 161.80 (CO) ppm; IR (KBr) ν: 3290, 3088, 2947, 2885, 2135, 1731, 1626, 1593, 1542, 1345, 1274, 1163, 1001, 962, 651 cm−1; MS (EI): m/z: 331.9 [M+].
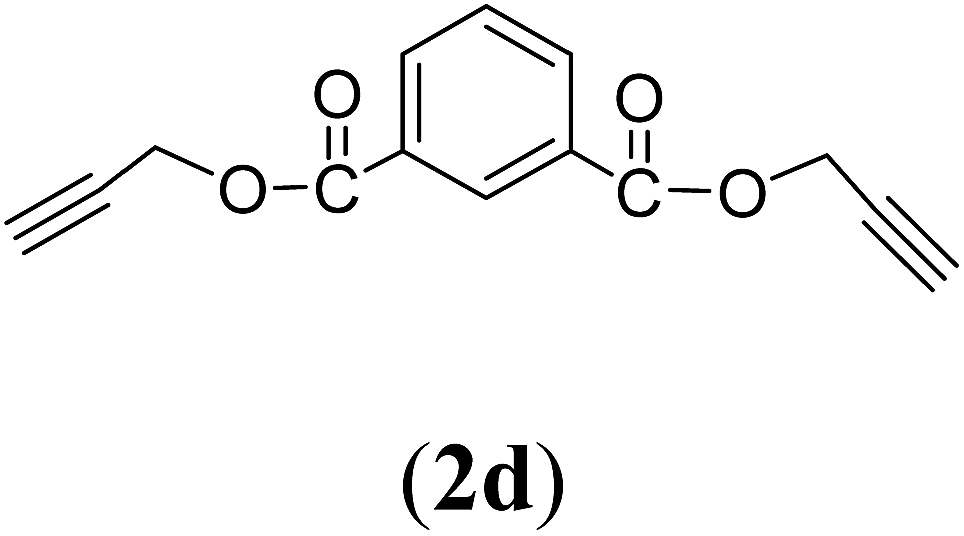
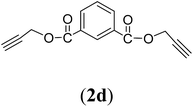
Bis(prop-2-ynyl)benzene-1,3-dicarboxylate (2d). 12.46 g, 85% yield; white solid; m.p. 85.53 °C; Rf: 0.78 (20% EtOAc/n-hexane); 1H-NMR (300 MHz, CDCl3) δ: 2.54–2.56 (t, J = 2.47 Hz, 2H, CH), 4.95–4.96 (d, J = 2.44 Hz, 4H, CH2), 7.56–7.59 (t, 1H, Ar), 8.25–8.29 (dd, J = 7.81, 1.73 Hz, 2H, Ar), 8.73 (s, 1H, Ar) ppm; 13C-NMR (300 MHz, CDCl3) δ: 53.57, 54.08 (CH2), 76.20, 77.31 (CH), 125.32, 130.15, 133.08, 134.05 (aromatic), 162.85, 163.94 (CO) ppm; IR (KBr) ν: 3265, 3086, 2943, 2874, 2129, 1716, 1607, 1236, 1084, 653 cm−1; GC-MS (EI, 70 eV) m/z: 242 [M+]; elemental anal. calcd (%) for C14H10O4: C, 69.42; H, 4.13; found: C, 69.10; H, 4.03.
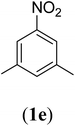
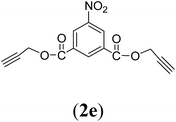
Bis(prop-2-ynyl)-5-nitrobenzene-1,3-dicarboxylate (2e). 3.9 g, 70% yield; white solid; m.p. 145.54 °C; Rf: 0.74 (20% EtOAc/n-hexane); 1H-NMR (300 MHz, CDCl3) δ: 2.59–2.61 (t, 2H, CH), 5.02–5.03 (d, J = 2.49 Hz, 4H, CH2), 9.02–9.08 (d, 3H, Ar) ppm; 13C-NMR (300 MHz, CDCl3) δ: 53.79 (CH2), 76.02, 77.67 (CH), 128.71, 131.92, 136.13, 148.51 (aromatic), 162.89 (CO); IR (KBr) ν: 3286, 3090, 2922, 2852, 2131, 1716, 1607, 1236, 1084, 653 cm−1; GC-MS (EI, 70 eV) m/z: 287 [M+]; elemental anal. calcd (%) for C14H9NO6: C, 58.53; H, 3.13; N, 4.87; found: C, 58.51; H: 3.06; N, 4.91.
General procedure for synthesis of bis(2-propynyloxy)benzene (4a–c)
A mixture of aromatic dihydroxybenzene (3a–c, 0.064 mol) and potassium carbonate (0.14 mol) was stirred in 60 mL dimethylformamide/acetone at 60 °C for 30 min. Then, propargyl bromide (0.15 mol) was added drop wise and the reaction mixture was heated at 60 °C for 5–10 h. The reaction progress was monitored by TLC. After the reaction was completed, the reaction mixture was cooled to room temperature and filtered. The filtrate was washed with water followed by brine. The extract was dried over sodium sulfate and concentrated on a rotary evaporator. A thick brown mass was collected and purified by column chromatography using 5% ethyl acetate in n-hexane.
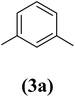
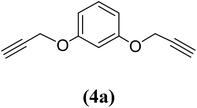
1,3-Bis(2-propynyloxy)benzene (4a). 7.74 g, 47% yield; amber colored solid; m.p. 40.69 °C; Rf: 0.82 (20% EtOAc/n-hexane); 1H-NMR (300 MHz, CDCl3) δ: 2.53–2.55 (t, J = 2.41 Hz, 2H, CH), 4.68–4.69 (d, J = 2.44 Hz, 4H, CH2), 6.62–6.65 (m, 3H, aromatic), 7.23 ppm (s, 1H, aromatic); 13C-NMR (300 MHz, CDCl3) δ: 55.97 (CH2), 75.80, 78.60 (CH), 102.56, 108, 130.10, 158.84 (aromatic) ppm; IR (KBr) ν: 3289, 3073, 2919, 2867, 2121, 1593, 1086, 682 cm−1; GC-MS (EI, 70 eV) m/z: 186 [M+]; elemental anal. calcd (%) for C12H10O2: C, 77.41; H, 5.37; found: C, 77.44; H, 5.28.
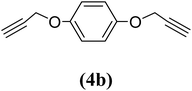
1,4-Bis(2-propynyloxy)benzene (4b). 9.33 g, 56% yield; cream coloured solid; m.p. 48.92 °C; Rf: 0.8 (20% EtOAc/n-hexane); 1H-NMR (300 MHz, CDCl3) δ: 2.52–2.53 (t, J = 2.41 Hz, 2H, CH), 4.65–4.66 (d, J = 2.38 Hz, 4H, CH2), 6.94 (s, 4H, aromatic) ppm; 13C-NMR (300 MHz, CDCl3) δ: 56.19 (CH2), 75.37, 78.65 (CH), 115.77, 152 (aromatic); IR (KBr) ν: 3289, 3049, 2921, 2864, 2120, 1593, 1031, 640 cm−1; GC-MS (EI, 70 eV) m/z: 186 [M+]; elemental anal. calcd (%) for C12H10O2: C, 77.41; H, 5.37; found: C: 77.29; H, 5.20.
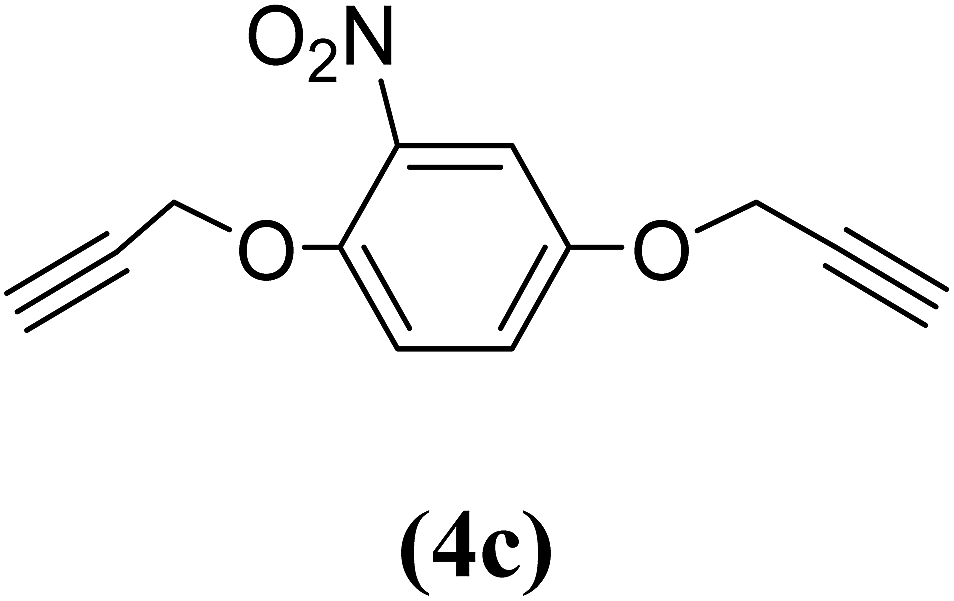
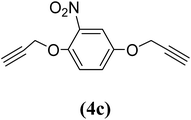
1,4-Bis(2-propynyloxy)-2-nitrobenzene (4c). 4.8 g, 46% yield; light yellow solid; m.p. 76.31 °C; Rf: 0.57 (20% EtOAc/n-hexane); 1H-NMR (300 MHz, CDCl3) δ: 2.57–2.58 (m, 2H, CH), 4.69–4.78 (m, J = 2.37 Hz, 4H, CH2), 7.19–7.23 (m, 2H, aromatic), 7.47 (d, 1H, aromatic) ppm; 13C-NMR (300 MHz, CDCl3) δ: 56.57, 58.05 (CH2), 76.45, 77.47 (CH), 111.60, 117.78, 121.24, 140.36, 145.45, 151.47 (aromatic) ppm; IR (KBr) ν: 3284, 3101, 2926, 2863, 2129, 1579, 1529, 1344, 1016, 650 cm−1; GC-MS (EI, 70 eV) m/z: 231 [M+]; elemental anal. calcd (%) for C12H9NO4: C, 62.33; H, 3.89; N, 6.06; found: C, 62.13; H, 3.80; N, 5.99.
General procedure for nitration of dicarboxylic acids
100 mL of oleum (18%) was transferred to a 500 mL three necked round bottomed flask with 75 mL fuming HNO3 (98%) mixed at a temperature below 25 °C. To this nitrating mixture 25 g of the corresponding dicarboxylic acid (1a, 1d)/p-toluic acid was added slowly maintaining the same temperature. The reaction mixture temperature was slowly raised to 90 °C and was maintained at that temperature for 4 h. Then the reaction mixture was cooled to room temperature and poured into 500 g of crushed ice and a white compound separated, was filtered, washed with water until free of acid. The product was purified by recrystallization from water to obtain white crystals.
Synthesis of 3,5-dinitrobenzene-1,4-dioic acid (1c)
40 mL of H2SO4 was transferred to a 250 mL three necked round bottom flask and 5 g (0.02 mol) 3,5-dinitro-p-toluic acid was added. To this reaction mixture 15 g (0.05 mol) sodium dichromate dihydrate was added in portions while maintaining the temperature between 35 and 40 °C. After completing the addition the reaction mixture was stirred for 2 h at 40 °C, kept overnight then poured into crushed ice (200 g). Cream colored product was separated, filtered and washed with cold water. The compound was purified by recrystallization from ethyl alcohol to obtain white crystals.
2-Nitrobenzene-1,4-dioic acid (1b). 27.2 g, 85% yield; white solid; m.p. 275 °C; 1H-NMR (300 MHz, DMSO-d6) δ, 7.95–7.97 (d, J = 7.93 Hz, 1H), 8.27–8.28 (dd, J = 7.93, 1.61 Hz, 1H), 8.37 (m, 1H); IR (KBr) ν: 3088, 1705, 1624, 1543, 1336 cm−1. Elemental anal. calcd (%) for C8H5NO6: C, 45.49; H, 2.36; N, 6.63; found: C, 45.11; H, 2.37; N, 6.52.
4-Methyl-3,5-dinitrobenzoic acid. 29.50 g, 88% yield; white solid; m.p. 159 °C; 1H-NMR (300 MHz, DMSO-d6), δ: 8.57 (s, 2H), 2.50 (s, 3H) ppm; IR (KBr) ν: 3082, 2877, 2831, 1703, 1631, 1541, 1350 cm−1. Elemental anal. calcd (%) for C8H6N2O6: C, 42.47; H, 2.65; N, 12.38; found: C, 42.38; H, 2.63; N, 12.38.
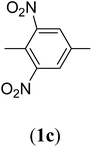
3,5-Dinitrobenzene-1,4-dioic acid (1c). 3.9 g, 69% yield; white solid; m.p. 279 °C; 1H-NMR (300 MHz, DMSO-d6) δ: 8.77 (s, 2H) ppm; IR (KBr) ν: 3099, 1714, 1622, 1546, 1346 cm−1. Elemental anal. calcd (%) for C8H4N2O8: C, 37.50; H, 1.56; N, 10.93; found: C, 37.26; H, 1.60; N, 10.73.
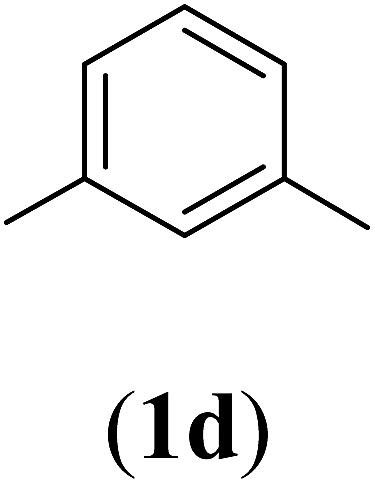
5-Nitrobenzene-1,3-dioic acid (1d). 21.17 g, 83% yield; white solid; m.p. 264 °C; 1H-NMR (300 MHz, DMSO-d6) δ: 8.66–8.69 ppm (d, 3H); IR (KBr) ν: 3545, 3462, 3007, 1703, 1626, 1541, 1352 cm−1.
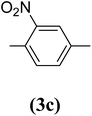
2-Nitrobenzene-1,4-diol (3c). 7.99 g, 60% yield; orange solid; m.p.: 133 °C; 1H-NMR (300 MHz, DMSO-d6) δ: 10.19 (s, 1H), 9.65 (s, 1H), 7.56 (d, 1H), 6.97–7.07 (m, 2H) ppm, (after D2O exchange: 7.05–7.16 (d, 1H), 6.74–6.92 (m, 2H) ppm; IR (KBr) ν: 3458, 3084, 1622, 1587, 1332 cm−1. Elemental anal. calcd (%) for C6H5NO4: C, 46.45; H, 3.22; N, 9.0; found: C, 46.21; H, 3.15; N, 8.93. GC-MS (EI, 70 eV) m/z: 155 [M+].
Acknowledgements
Authors thanks Director, HEMRL, Pune, India for his interest on this study and providing infrastructure resources.
References
- T. Keicher, K. Werner, E. Siegfried, W. Tim, K. Manfred and K. Horst, Int. Annual Conference of ICT, June 29–July 2, 2010, pp. 12/1–15 Search PubMed.
- S. K. Reshmi, K. P. Vijayalakshmi, D. Thomas, E. Arunan and C. P. Reghunadhan Nair, Propellants, Explos., Pyrotech., 2013, 38, 525–532 CrossRef CAS.
- M. Klaus, H. Thomas, S. Wenka, T. Keicher and K. Horst, Propellants, Explos., Pyrotech., 2009, 34, 218–230 CrossRef.
- R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565–598 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- R. N. Warrener, R. A. Russell and S. M. Marcuccio, Aust. J. Chem., 1980, 33, 2777–2990 CrossRef CAS.
- A. R. Katritzky, Z. Yuming, S. K. Singh and J. S. Peter, ARKIVOC, 2003,(xv), 47–64 CAS.
- A. P. Manzara and L. E. Minn, U S. Pat., 5681904A, 1997.
- S. M. Byoung, C. P. Young and J. C. Yoo, Propellants, Explos., Pyrotech., 2012, 37, 59–68 CrossRef.
- E. Landsem, L. J. Tomas, T. E. Kristensen, K. H. Finn, B. Tore and U. Erik, Propellants, Explos., Pyrotech., 2013, 38, 75–86 CrossRef CAS.
- H. Potinus, A. B. Manfred and A. Jasmin, Int. Annual Conference of ICT, June 24–27, 2008, pp. 129/1–34 Search PubMed.
- J. H. Wotiz, US Pat., 2, 979, 538, 1961.
- V. Haridas, K. Lal, Y. K. Sharma and S. Upreti, Org. Lett., 2008, 10, 1645–1647 CrossRef CAS PubMed.
- R. X. Yao, L. Kong, Z. S. Yin and F. L. Qing, J. Fluorine Chem., 2008, 129, 1003–1010 CrossRef CAS.
- M. M. Osama, L. M. Sridhar and W. Y. H. Qingwen, WO2008048733 A1, 2008.
- M. Ghaemy and H. Mighani, Chin. Chem. Lett., 2009, 20, 800–804 CrossRef CAS.
- A. Blatt, J. Org. Chem., 1960, 25, 2030–2034 CrossRef CAS.
- B. Yin, G. Huang and J. Pan, CN 101219958 A, 2008.
- L. M. Zenon and L. S. Cynthia, US Pat., 5,099,057, 1992.
- G. A. How and P. Sreekumar, Energetic Polymers, Wiley-VCH Verlag & Co KGaA, Germany, 2012 Search PubMed.
- Thermo Scientific OMNIC Spectra Software-Application note 51773 Search PubMed.
- M. A. Escola, C. A. Moina, A. C. Nino Gomez and G. O. Ybarra, Polym. Test., 2005, 24, 572–575 CrossRef CAS.
- A. Osei-Owusu and G. C. Martin, Polym. Eng. Sci., 1991, 31, 1604–1609 CAS.
- K. Husnu, F. Petel and S. J. Ozkar, Appl. Polym. Sci., 2001, 80, 65–70 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: All the spectral data for characterization of the compounds are provided. See DOI: 10.1039/c5ra25909f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.