
Improved power density of an enzymatic biofuel cell with fibrous supports of high curvature†
 *e
and
*e
and
Abstract
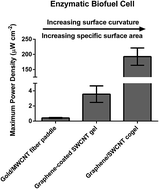
Enzyme immobilization onto gold- or carbon nanotube-based nanomaterials has driven recent advances in the development of enzymatic biofuel cells (EBFCs). Enzyme–gold and enzyme–carbon nanotube interfaces are conducive to achieving efficient electron transfer between the enzyme active site and an electrode along with high enzyme loadings. Herein, we investigate the performance dependence of EBFCs on the surface curvature, specific surface area (SSA) and pore size of underlying enzyme supports. One of the supports was gold/multi-wall carbon nanotube (MWCNT) fiber paddles that were formed by depositing gold nanoparticles and MWCNTS onto electrospun polyacrylonitrile fibers with a diameter of 1 μm and a SSA of 3.6 m2 g−1 with micrometer sized pores. The other support was graphene-coated single-wall carbon nanotube (SWCNT) gels, which had 1 nm thick struts, 686 m2 g−1 SSA, and pores of diameter ≤ 15 nm. Glucose oxidase (GOX) and bilirubin oxidase (BOD) were immobilized onto each material to form enzymatically active anodes and cathodes, respectively. EBFCs constructed using gold/MWCNT fiber paddle electrodes yielded power densities of 0.4 μW cm−2 with an open circuit voltage of 0.22 V and GOX loadings of 2.0 × 10−10 mol cm−2. In comparison, EBFCs utilizing graphene-coated SWCNT gel electrodes had 10-fold lower GOX loadings (1.0 × 10−11 mol cm−2), but still produced 10-fold greater power densities (≈3.6 μW cm−2) and an open circuit voltage of 0.22 V. We postulate that a greater fraction of GOX supported on graphene-coated SWCNTs that had high curvature retained their biochemical functionality. Further, this study provides a design principle for improving enzymatic power generation.
 Please wait while we load your content...
Please wait while we load your content...

