
Anionic hexadeca-carboxylate tetrapyrazinoporphyrazine: synthesis and in vitro photodynamic studies of a water-soluble, non-aggregating photosensitizer†
Miloslav Machaceka,
Jan Kollárb,
Miroslav Miletinb,
Radim Kučerab,
Pavel Kubátc,
Tomas Simuneka,
Veronika Novakova*d and
Petr Zimcik*b
aDepartment of Biochemical Sciences, Faculty of Pharmacy in Hradec Kralove, Charles University in Prague, Heyrovskeho 1203, 50005, Hradec Kralove, Czech Republic
bDepartment of Pharmaceutical Chemistry and Pharmaceutical Analysis, Faculty of Pharmacy in Hradec Kralove, Charles University in Prague, Heyrovskeho 1203, 50005, Hradec Kralove, Czech Republic. E-mail: zimcik@faf.cuni.cz; Tel: +420 495067257
cJ. Heyrovský Institute of Physical Chemistry, v.v.i., Academy of Sciences of the Czech Republic, Dolejškova 3, 182 23 Praha 8, Czech Republic
dDepartment of Biophysics and Physical Chemistry, Faculty of Pharmacy in Hradec Kralove, Charles University in Prague, Heyrovskeho 1203, 50005, Hradec Kralove, Czech Republic. E-mail: veronika.novakova@faf.cuni.cz; Tel: +420 495067380
First published on 15th January 2016
Abstract
A sodium salt of zinc tetrapyrazinoporphyrazine bearing eight 3,5-dicarboxylatophenyl substituents (1) was synthesized. The presence of sixteen negative charges in a rigid arrangement on the periphery of the macrocycle inhibited its aggregation in water or buffers at pH > 5.8. Strong aggregation was, however, observed in buffers at pH < 4.8 due to the protonation of carboxylate functions. Fluorescence microscopy revealed that the compound localized to lysosomes and endosomes in cells. The compound's photodynamic activity on HeLa cancer cells (IC50 = 5.7 ± 1.1 μM) was found to be influenced by both pH and interactions with serum proteins. This was demonstrated with a detailed in vitro study based on the inhibition of vacuolar H+-ATPase using bafilomycin A1, which increased the intracellular fluorescence of 1. Compound 1 also formed interactions with serum proteins that partially quenched its excited states; however, they also protected the compound from self-aggregation at low pH.
Introduction
Phthalocyanines (Pcs) and their aza-analogues (e.g., tetrapyrazinoporphyrazines (TPyzPzs)) represent an interesting group of organic dyes with diverse uses in different areas of research and practical applications.1 One key application is in medicine, specifically in photodynamic therapy (PDT) to treat cancer. PDT involves the combination of three components: a photosensitizer (PS), light and oxygen.2–4 The energy of the light that is absorbed by the PS is transferred to a ground state triplet oxygen, forming singlet oxygen. This highly reactive species subsequently destroys specific subcellular compartments; depending on the localization of the PS, this can lead to cell death.5 The main drawbacks associated with using Pcs in PDT include their low solubility in water and the strong tendency of these planar compounds with hydrophobic cores to aggregate. For this reason, a number of water-solubilizing substituents (cationic, anionic or non-ionic) have been attached to the periphery of Pcs.6 Although water-soluble Pcs have been developed, many of them aggregate in water, preventing them from achieving high photodynamic activity. Pcs that are both water-soluble and non-aggregating in the absence of surfactants or disaggregating additives are rare, and few have been reported to date.7–9 Among the most successful aggregation-inhibiting strategies in water-based media are axial substitutions on the central metal10,11 or substitutions with multiple charged peripheral substituents directly attached to the core12 or in a rigid arrangement.7–9TPyzPzs are promising aza-analogues of Pcs that have garnered increasing attention in several emerging applications,13 including use as fluorescent sensors14 or as dark quenchers of fluorescence in DNA hybridization probes.15 The photophysical properties that are important for PDT (e.g., singlet oxygen and fluorescence quantum yields) are almost identical to the key features of Pcs, making their use in PDT an interesting prospect.16 However, at the time of this writing, only two studies reporting biological evaluations of TPyzPzs could be found.2,17
This work is focused on the synthesis and photodynamic evaluation of the first reported anionic zinc TPyzPzs with eight 3,5-dicarboxylatophenyl substituents (1, Fig. 1). The design of sixteen charged substituents in rigid arrangements was created to fully inhibit aggregation in water by way of electrostatic repulsion forces. Indeed, a similar approach using rigid cationic substituents (Pc-Im16, Fig. 1) with stable charges was recently reported to result in a fully non-aggregating zinc Pc in water, which showed high photodynamic activity in vitro.7 Therefore, we were interested in investigating anionic derivatives with rigid peripheral arrangements and comparing them to the highly active cationic Pc-Im16.
 | ||
| Fig. 1 Structures of the investigated compounds. | ||
Results and discussion
Synthesis
TPyzPzs are typically prepared from corresponding pyrazine-2,3-dicarbonitriles that result from the condensation of diaminomaleonitrile (DAMN) with suitably substituted vicinal diketones.18 According to published procedures, the starting material trimesic acid was esterified to triester 3 (ref. 19) that was partially hydrolyzed to diester 4 (ref. 20) with a subsequent reduction of the carboxylic function to alcohol 5 (ref. 19) followed by its selective oxidation to aldehyde 6 (ref. 21) (Scheme 1). Aldehyde 6 was subjected to benzoin condensation and afforded the corresponding acyloin. However, the acyloin was unstable and gradually oxidized to a diketone during purification and chromatography on silica. For this reason, it was directly converted to diketone 7. Substantial differences in the yields of 7 were observed when using different conditions for the first step, i.e., the benzoin condensation. When KCN was used as a catalyst in DMSO at 135 °C, it only led to an 18% yield of the diketone (after oxidation), and no improvement was made by changing the solvent (EtOH, BuOH, DMF, or pyridine) or temperature from room temperature to reflux. A substantial increase in the yield of compound 7 to 77% was achieved by changing the catalyst to 5-(2-hydroxyethyl)-3,4-dimethylthiazolium iodide. Subsequent condensation of diketone 7 with DAMN created pyrazine 8 with a yield of 76%.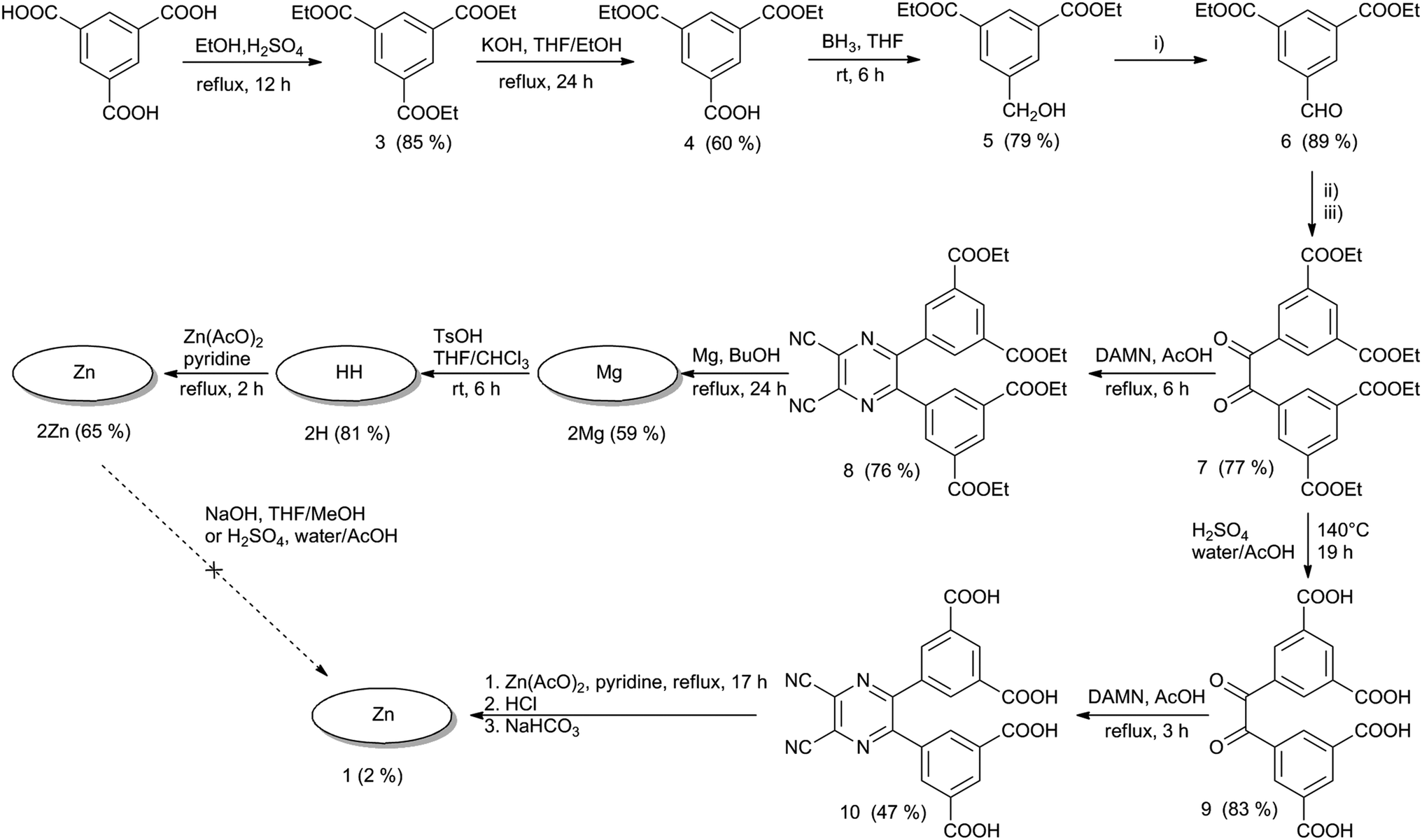 | ||
| Scheme 1 Synthesis of target compounds. Reaction conditions: (i) pyridinium chlorochromate, CHCl3, rt, 6 h; (ii) 5-(2-hydroxyethyl)-3,4-dimethylthiazol-3-ium iodide, DBU, EtOH, reflux, 3 h; (iii) Cu(AcO)2, NH4NO3, AcOH, reflux, 2 h. | ||
Cyclotetramerization of pyrazine 8 induced by magnesium butoxide was used to synthesize the magnesium TPyzPz 2Mg. Concomitantly, complete transesterification from ethyl to butyl esters was observed (see MS spectra in Fig. S18†) in accordance with similar, previously reported reactions.9 Treatment of the magnesium complex with p-toluenesulfonic acid (TsOH) gave metal-free derivative 2H, which was complexed with zinc using zinc acetate in pyridine. Zinc complexes are known for much higher singlet oxygen quantum yields than their magnesium counterparts.22 Interestingly, 2H gave very complex NMR spectra where all the signals were doubled or even multiplied (Fig. S5 and S6†). On the other hand, MS spectra indicated only a correct mass of the product (with one fragment due to loss of C2H4 caused by ionization) and fully satisfactory elemental analysis. It suggests that the origin of the spectra complexity can be in specific supramolecular arrangement of two (or more) TPyzPzs molecules, similarly as observed e.g. for formation of J-dimers for zinc complexes.23 In this case, however, the central hydrogens may be involved in H-bonding as also indicated by no splitting in absorption spectra (see below).
All three derivatives of TPyzPz 2 were highly soluble in organic solvents but not in water. Attempts to hydrolyze the ester groups in 2Zn were unsuccessful. Basic hydrolysis using hydroxides, a process that is often successfully reported for Pcs,9,24 led to decomposition of the TPyzPz macrocycle. The reaction mixture turned yellow without any absorption in the Q-band region as indicated by UV-vis spectroscopy. Similarly, reports of decreased TPyzPz stability in the presence of strong bases can be found.25 Acidic hydrolysis using H2SO4 did not lead to complete conversion even after prolonged reaction time and increased temperature. The failure of acidic hydrolysis was indicated by insolubility of the product in basic water (pH > 8).
As an alternative synthetic route to 1, we attempted to hydrolyze ester groups in pyrazine 8, but very complex mixtures of products were obtained after both basic and acidic hydrolyses. The likely explanation for this result is that CN groups in 8 were also hydrolyzed at the same time. Finally, treatment of diketone 7 with H2SO4 afforded the corresponding diketone 9, which had free carboxylic groups. The condensation of 9 with DAMN led to pyrazine 10 with a 47% yield (Scheme 1). A number of methods to cyclotetramerize this pyrazine (including reactions with magnesium or lithium butoxides, reactions induced by DBU in BuOH and a template method using zinc acetate in quinoline or DMF) were attempted, but the only reaction that led to a green product was a template method that used zinc acetate in pyridine. The green solid from this reaction was insoluble in all tested solvents, most likely as a consequence of the formation of zinc salts on the peripheral carboxylic functions. The free carboxylic functions were released by treatment with HCl and then converted to hexadeca sodium salt 1 with a slight excess of NaHCO3. TPyzPz 1 was well soluble in water and was purified by gel filtration on Sephadex.
Absorption spectra
The absorption spectra of esters 2Mg, 2H and 2Zn in pyridine consisted of two important bands: a low energy Q-band over 650 nm and a high energy B-band at approximately 375 nm (Fig. 2a). The sharp Q-band shape typical for monomeric species was observed for all three compounds in pyridine, including metal-free 2H, which was expected to be split due to reduced symmetry. In this particular case, the lack of Q-band splitting can be explained by the deprotonation of central NH in a basic solvent and the formation of a “proton-transfer complex” in a manner similar to those reported for other Pcs and TPyzPzs.26 The spectra of 2Mg and 2Zn in other solvents (THF, toluene) showed a wide Q-band shape and the appearance of additional bands in this area, indicating the presence of aggregation (Fig. S25†). Interestingly, the sharp shape of the absorption spectrum of 2H in THF or toluene, qualitatively different than that observed in pyridine, did not indicate aggregation. The unsplit character in these non-basic solvents suggested that central hydrogens responsible for decreased symmetry of the molecule might be somehow removed, e.g. by forming H-bonds with neighboring molecule as suggested already by complex NMR spectrum.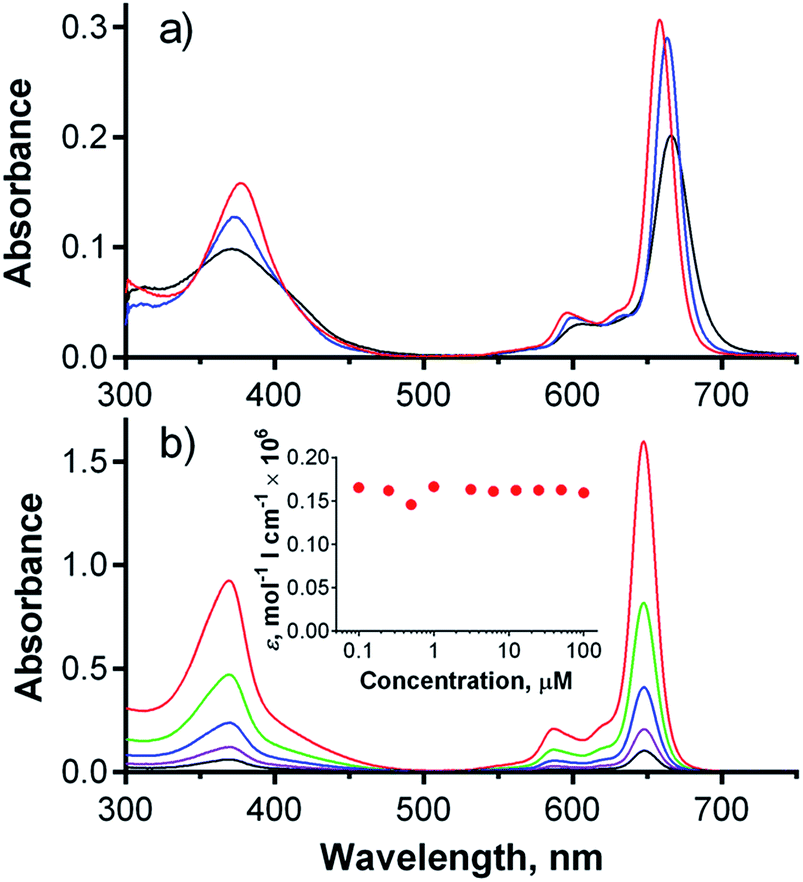 | ||
| Fig. 2 (a) Absorption spectra of 2Mg (blue), 2H (black) and 2Zn (red) in pyridine at a concentration of 1 μM. (b) Absorption spectra of 1 in phosphate–citrate buffer (pH 7.1) at concentrations ranging from 6 μM (black) to 100 μM (red) as determined in cuvette with a 1 mm path length. Inset: dependence of the molar extinction coefficient of 1 on concentration in phosphate–citrate buffer (pH 7.1). | ||
The absorption spectra of hexadeca sodium salt 1 in water corresponded to the monomeric form (Fig. S26†). The well-resolved shape of the Q-band was also retained at high concentration (100 μM) in phosphate–citrate buffer (pH 7.1) and had a constant molar extinction coefficient over a wide range of concentrations (Fig. 2b, inset). Importantly, the monomeric character was also retained in serum-free cell culture medium (SFM), suggesting that the compound is essentially in monomeric form, even in the presence of a complex mixture of ions. Such a low tendency for aggregation in aqueous medium is rare among zinc complexes of Pcs and TPyzPzs, and very few similar reports can be found.7,9 The full inhibition of aggregation was most likely a consequence of the rigid arrangement of the peripheral phenyl substituents directly attached to the core; this arrangement forced the carboxylate functions to assume positions above and below the TPyzPz plane. The presence of negative charges may fully inhibit any TPyzPz molecules from approaching each other as a result of electrostatic repulsion forces. The rigid arrangement seems to be of crucial importance for efficient inhibition of aggregation, as similar anionic Pcs bearing carboxylates on flexible linkers still aggregate in water,11 even those that contain 32 carboxylate functions on dendrons.27
Photophysical characterization
The fluorescence emission spectra of all of the studied TPyzPzs (in pyridine for 2Mg, 2H and 2Zn and in water for 1) were typical for this type of compound mirroring the Q-band shape (Fig. S26†) with small Stokes shift that did not exceed 10 nm (Table 1). The excitation spectra (Fig. S26†) perfectly matched the absorption spectra, further confirming that no aggregates were present and that the compounds existed exclusively in monomeric form in solution. Determination of photophysical parameters was therefore not influenced by aggregation.| Compound | Solvent | λmax/nm | λF/nm | ΦFb | ΦΔb |
|---|---|---|---|---|---|
| a Absorption maximum (λmax), fluorescence maximum (λF), fluorescence quantum yield (ΦF), singlet oxygen quantum yield (ΦΔ).b Using ZnPc as a reference (ΦF(THF) = 0.32, ΦΔ(pyridine) = 0.61), the reported values are the mean of three independent measurements with an estimated error of ±15%.c ΦF(1) = 0.36 in buffer (pH 7.1).d In D2O, using methylene blue as a reference (ΦΔ(D2O) = 0.52). Other photophysical data for 1 in D2O: triplet state lifetime τT = 677 μs (in argon-saturated solution), rate constant for triplet state quenching by oxygen kO2 = (1.40 ± 0.06) × 109 M−1 s−1.e ΦΔ(1) = 0.26 in D2O 0.1 M buffer (pD = 7.1). | |||||
| 1 | Water | 647 | 657 | 0.31c | 0.23d,e |
| 2Mg | Pyridine | 663 | 672 | 0.53 | 0.37 |
| 2H | Pyridine | 666 | 671 | 0.13 | 0.29 |
| 2Zn | Pyridine | 659 | 667 | 0.27 | 0.58 |
Singlet oxygen quantum yields (ΦΔ) and fluorescence quantum yields (ΦF) were determined using comparative methods with unsubstituted ZnPc as a reference compound and 1,3-diphenylisobenzofuran (DPBF) as a singlet oxygen trap (Table 1). Of the esters of 2, the fluorescence emission was more intense for 2Mg while singlet oxygen production was highest for 2Zn, which is in full agreement with the heavy atom effect.22 With respect to PDT, the zinc complex seems to be of interest due to its high ΦΔ values (ΦΔ(2Zn) = 0.58). The low ΦΔ and ΦF of 2H are typical for metal-free TPyzPzs, although this phenomenon has not yet been fully explained.28 Interestingly, anionic TPyzPz 1 was characterized by intense red fluorescence in water (ΦF = 0.31) and also in a buffer at pH 7.1 (ΦF = 0.36), indicating that it is in an active form even in water-based media. Time-resolved near-infrared luminescence of singlet oxygen O2(1Δg) at 1270 nm was used to determine ΦΔ values of 1 in water because the chemical trap (DPBF) is not soluble in this solvent. The values of ΦΔ were 0.23 in D2O and 0.26 in buffer. A decrease of ΦΔ in aqueous solution compared to organic solvent is a common phenomenon both for approved PDT photosensitizers and for those in clinical trials.4 As mentioned above, the aggregation of 1 can be excluded; therefore, other solvent-induced relaxation processes and lower oxygen solubility in water may account for the lower values. Similar to our results, a fully non-aggregating zinc Pc containing hexadecacationic imidazol substituents in a rigid arrangement (Pc-Im16) was reported to have a lower ΦΔ value in D2O versus organic solvent.7
In vitro evaluation of photodynamic activity
As shown above, compound 1 had favorable photophysical parameters that warranted its consideration as a PS for PDT. Furthermore, it must be noted that this compound can be applied to cells without the addition of any co-solvents (e.g., DMSO, DMF) or surfactants (e.g., cremophor) that are typically used for in vitro evaluation of Pcs due to the low solubilities and/or aggregation tendencies of such compounds in water environment.The first in vitro experiment investigated the rate of cellular uptake of 1 by a human cervix carcinoma cell line (HeLa). As shown in Fig. 3a, the uptake of 1 by cells was rapid within the first 1–2 hours and then significantly slowed, reaching a plateau with a final amount of 0.20 nmol of 1 per mg of protein. This cellular uptake profile is in good agreement with those produced by similar anionic zinc Pcs substituted with eight 3,5-dicarboxylatophenoxy substituents.9 For comparison, the non-aggregating cationic Pc-Im16 had 2.5-fold higher uptake with approximately 0.55 nmol mg−1 of protein (Fig. 3a).
 | ||
| Fig. 3 (a) Cellular uptake of compound 1 (red, dots) and Pc-Im16 (blue, open squares) by HeLa cells after incubation with a 4 μM concentration of each dye. (b) Dark toxicity (black triangle) and phototoxicity (red dots, λ > 570 nm, 11.2 J cm−2) of 1 in HeLa cells. Typically, four independent experiments, each in quadruplicate, were performed. Key: *, p < 0.05; **, p < 0.01; ***, p < 0.001. | ||
Subsequently, toxicity and photodynamic activity were assessed using HeLa cells. Dark toxicity (i.e., without activation by light) was low (half-maximum toxic concentration, TC50 = 154 ± 8.5 μM). Activation of the compound with light (λ > 570 nm, 11.2 J cm−2) induced a lethal photodynamic effect in HeLa cells with a half-maximum inhibitory concentration (IC50) of 5.7 ± 1.1 μM (Fig. 3b). This value is comparable with data reported for similar anionic zinc Pcs substituted with eight slightly more flexible 3,5-dicarboxylatophenoxy substituents, which had an IC50 > 4 μM in HepG2 cells (λ > 600 nm, 48 J cm−2)24 and an IC50 = 4.5 μM in HEp2 cells (λ > 610 nm, 1 J cm−2).9 It is also comparable to other carboxy-substituted Pcs (see Table S1†), suggesting that 1 might be a useful PS for PDT. However, octacationic zinc Pcs with flexible triethylammonioethylsulfanyl substituents attached to their cores in the α-(non-peripheral) or β-(peripheral) positions were previously shown to be considerably more active on HeLa cells, with IC50 values of 0.31 μM and 0.54 μM, respectively (λ > 570 nm, 11.2 J cm−2).2 An even lower IC50 of 0.037 μM was obtained for Pc-Im16 on HeLa cells (λ > 570 nm, 11.2 J cm−2).7 In consideration of the published data on cationic Pcs, an IC50 of 5.7 ± 1.1 μM for compound 1 suggests a rather modest photodynamic activity. It should be noted that compound 1 is fully monomeric in water even at high concentrations. This non-aggregating behavior in water resembles cationic Pc-Im16, which can be used for comparison due to its equal number of charged substituents, which are also in a rigid arrangement.7 The large difference in photodynamic activities (two orders of magnitude) between anionic 1 and cationic zinc Pc-Im16 cannot be explained only by the slightly lower ΦΔ value (0.26) of 1 in D2O buffer (compared to a ΦΔ value of 0.33 for Pc-Im16 in D2O) and the 2.5-fold higher uptake of Pc-Im16 (see above).7 To explain these observations, the fate of compound 1 in cells was studied in greater detail.
Subcellular localization
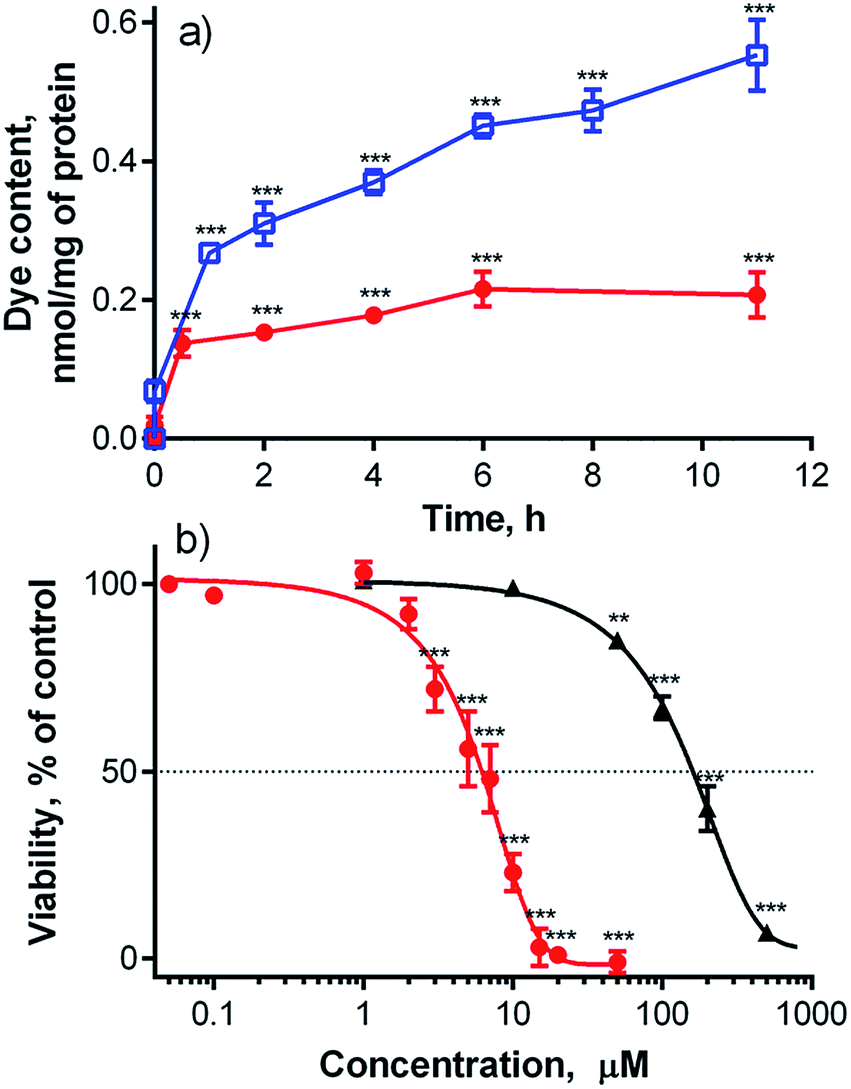
The subcellular localization of 1 was assessed using fluorescent probes specific for distinct organelles; these included MitoTracker Green for mitochondria and LysoTracker Blue for lysosomal compartments. Compound 1 produced red punctate fluorescence throughout the whole cytoplasm and showed no overlap with the green-stained mitochondria (Fig. S30†). Considering the highly charged anionic character of 1 and its punctate fluorescence distribution, its localization in endosomes/lysosomes was expected. However, no LysoTracker Blue signal was visible upon co-incubation with 1. One plausible explanation for this finding is that LysoTracker Blue, which is basic and protonated in lysosomes, formed a ground-state complex with anionic 1, leading to static quenching of the fluorescence. This assumption was confirmed by the changes in absorption spectra and fluorescence intensities (Fig. S27 and S28†) that were observed upon mixing solutions of 1 and LysoTracker Blue. The absorption spectrum of the mixture did not correspond to the simple sum of the spectra of the individual components, indicating the formation of a ground-state heterodimer that is non-fluorescent, as is typical for such interactions.29 This was supported by the substantial decreases in the fluorescence intensities of both LysoTracker Blue and compound 1 (Fig. S27†). From this, we concluded that the use of basic probes for lysosomes may not be suitable for the visualization of anionic TPyzPzs when inside these organelles. For this reason, dextran labeled with fluorescein (a rather acidic, organelle-specific probe) was used for visualization of endo-lysosomal vesicles. This probe is less selective and, in addition to lysosomes, can be used to visualize endosomes, which are not as acidic as lysosomes. When administered to cells, the red signal of compound 1 and the green signal of the probe colocalized in a punctate pattern throughout the whole cytoplasm (Fig. 4).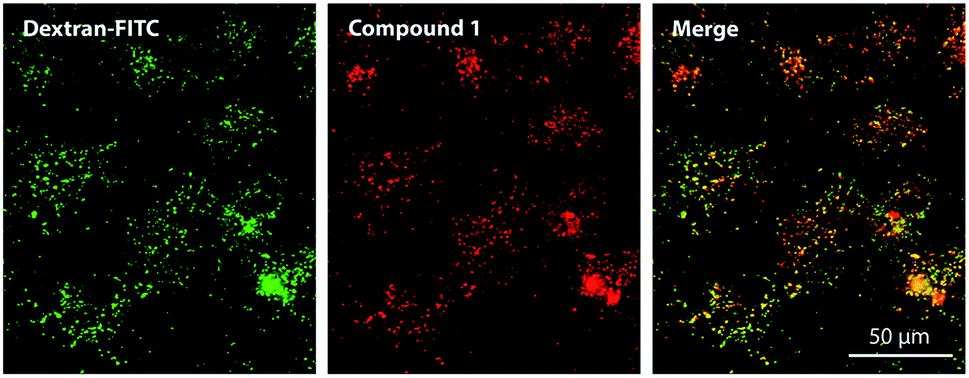 | ||
| Fig. 4 Subcellular localization of compound 1 (red) in HeLa cells as visualized by fluorescence microscopy after co-incubation with fluorescein-labeled dextran (green). | ||
A significant population of compound 1-positive vesicles was found in the perinuclear area. Such vesicles are often organelles with rather lower internal pHs, such as lysosomes (pH 4.5–5) and endosomes (pH 5.5–6.5).30 However, staining with fluorescein-labeled dextran cannot distinguish different types of endo-lysosomal vesicles. To determine the specific localization of the fluorescent signal corresponding to 1, cell transfection with fluorescent proteins using baculovirus-mediated gene delivery was performed. Lysosomes were labeled with red fluorescent protein (RFP) fused to lysosomal-associated membrane protein 1 (Lamp1),31 and endosomes were labeled with green fluorescent protein (GFP) fused to Rab7a32 (Fig. 5). Major population of red signal of 1 was colocalized with both Lamp1 and Rab7a, suggesting that it originates from both lysosomes and endosomes with comparable intensity. Positivity of lysosomes for both proteins was also detected during the experiments (Fig. S31†). This was not an artifact of the transfection or the imaging procedure, as the association of Rab7 with lysosomes33 has already been described in literature.34
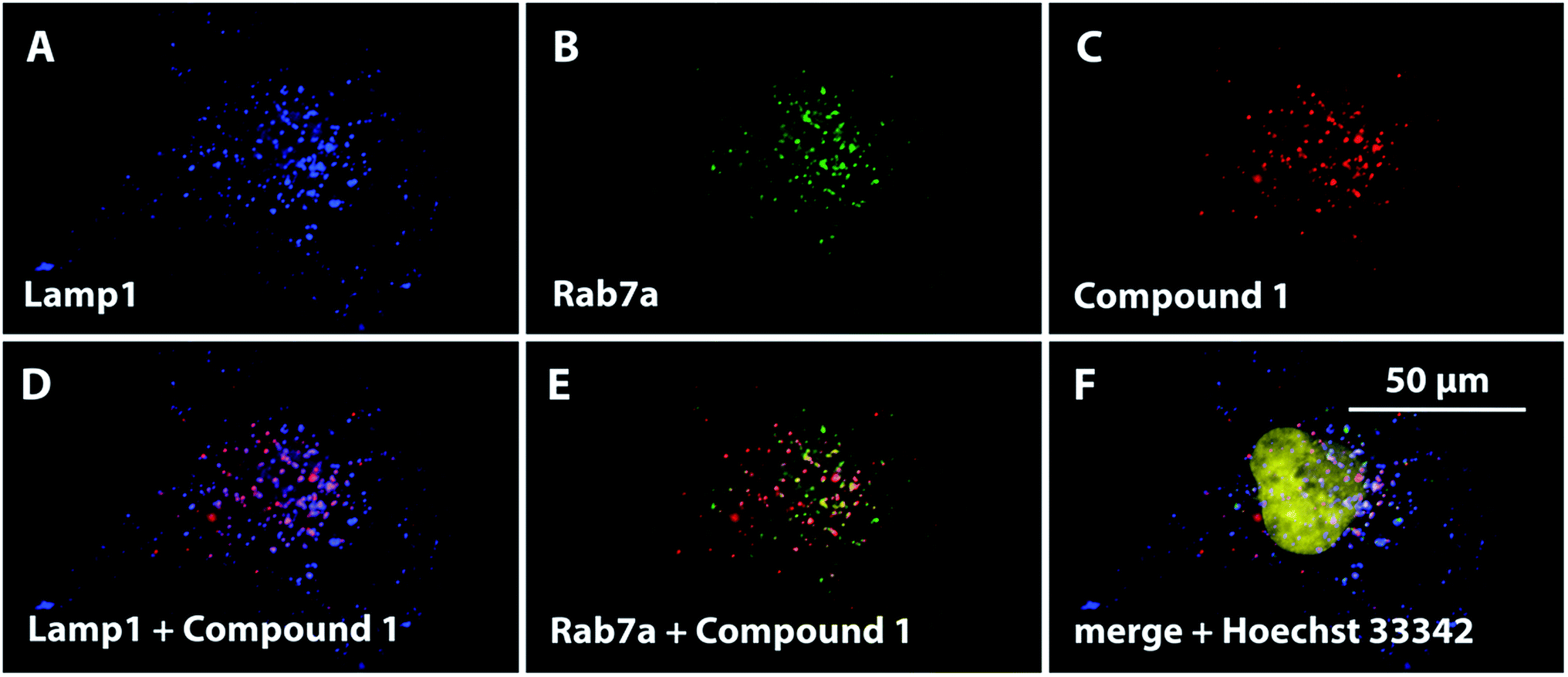 | ||
| Fig. 5 A pseudocolored visualization of the subcellular localization of Lamp1-RFP (blue, A), Rab7a-GFP (green, B) and compound 1 (red, C) in HeLa cells. The colocalization of 1 with both labeled proteins is displayed as the merged microphotographs (D, E); the nucleus was stained with Hoechst 33342 (yellow) and its position was visualized in a merged microphotograph (F). | ||
Influence of pH
Given the localization of anionic 1 in acidic organelles, the photophysical properties of 1 were examined as a function of buffer pH. Strong fluorescence (ΦF = 0.36) was observed at pH > 5.7, and a complete loss of signal occurred at pH < 4.8 (Fig. 6, inset). The fluorescence decrease at low pH was accompanied by changes in the absorption spectra that are typical of the formation of aggregates (Fig. 6). This suggests that compound 1 is non-aggregated and active in its fully ionic form at high pH and that protonation of some of its carboxylates occurs below pH 5.7. Without their negative charges, the carboxyl functions no longer promote a monomeric state in water, leading to extensive aggregation that renders the compound photodynamically inactive. Interestingly, the distinct changes in absorption spectra could potentially be used for future development of a wavelength-ratiometric probe for fine measurements of pH in the range of 4.8 to 5.6 (Fig. S29†); however, such work is beyond the scope of this paper.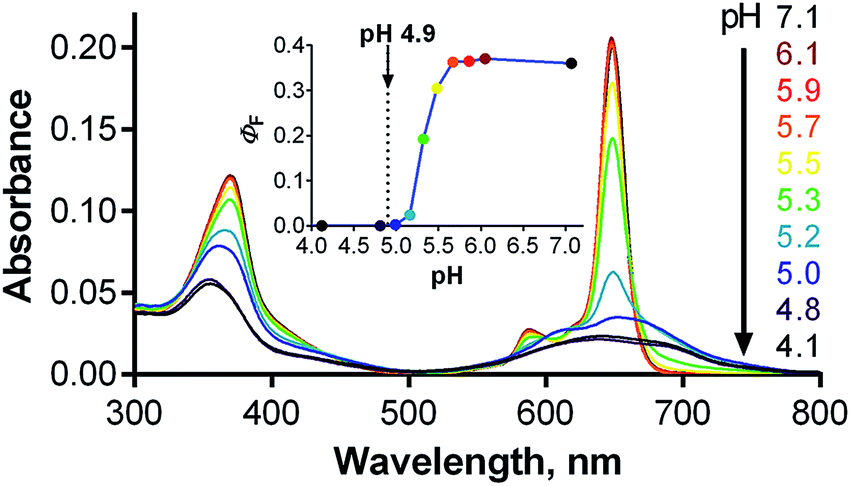 | ||
| Fig. 6 Changes in the absorption spectra of 1 (c ∼ 1 μM) in phosphate–citrate buffer at different pHs. Inset: dependence of ΦF of 1 on buffer pH (λexc = 594 nm). The arrow indicates the average lysosomal pH in HeLa cells as determined in this work. | ||
The average lysosomal pH in HeLa cells was measured using LysoSensor Yellow/Blue DND-160 (Fig. S32†). The obtained pH value of 4.9 is concordant with the reported pH range (4.5–5.0) for lysosomes.30 At this pH, compound 1 should be in an aggregated (i.e., non-fluorescent) state in lysosomes (Fig. 6, inset). Therefore, an increase in lysosomal pH would be expected to support monomerization and subsequently significantly increase the fluorescence signal. Bafilomycin A1 is a selective inhibitor of vacuolar type H+-ATPase (V-ATPase), which is reported to increase lysosomal pH above 6.0 in living cells after few hours of treatment.35 Therefore, HeLa cells were treated with compound 1 and bafilomycin A1, and the fluorescence signal of 1 was monitored as a function of time following the inhibitor treatment. After three hours, the fluorescence signal at 671 nm almost doubled (Fig. 7). Nearly identical results (increase in fluorescence to 183% of control intensity) were obtained in experiments using 20 mM NH4Cl, another agent capable of increasing lysosomal pH (Fig. S33†).36 For comparison, no significant changes in fluorescence were observed upon treatment of HeLa cells with cationic Pc-Im16 after co-incubation with bafilomycin A1 (Fig. 7) or NH4Cl. This is in accordance with the reported behavior of this cationic compound, which was found to retain its monomeric state regardless of buffer pH.7
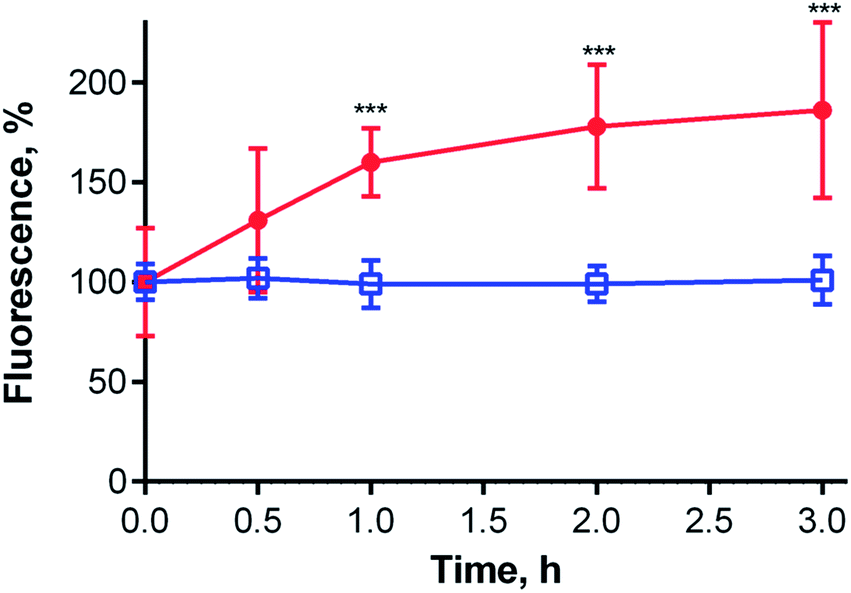 | ||
| Fig. 7 Changes in the fluorescence intensity of compound 1 (red dots; λexc = 375 nm, λem = 671 nm) and Pc-Im16 (blue open squares; λexc = 363 nm, λem = 693 nm) in HeLa cells as a function of incubation time with bafilomycin A1. Two independent experiments, each with 10 replicates, were performed. Key: *, p < 0.05; **, p < 0.01; ***, p < 0.001. | ||
The above experiments demonstrated that lysosomal pH may play a role in the activity of 1; however, they did not unequivocally explain the behavior of this compound. Considering the data presented in Fig. 6, the fluorescence signal of 1 in lysosomes should be barely detected (ΦF(1) < 0.001 in buffer at pH 4.9). However, compound 1 was well visualized in lysosomes by fluorescence microscopy (Fig. 4 and 5), and its signal in lysosomes was of comparable intensity to its signal in endosomes, which have a typical pH of 5.5–6.5. The fluorescence signal of 1 in endosomes was expected to be much stronger than that in lysosomes (ΦF(1) = 0.30–0.35 in buffer at pH 5.5–6.5). At the same time, increasing the lysosomal pH using bafilomycin A1 led to only a twofold enhancement of the fluorescence, which is not in agreement with the anticipated increase, which was expected to be at least one order of magnitude. This disparity suggests that pH is not the only factor influencing the photodynamic activity of 1.
Interactions of the studied compounds with serum proteins
A number of studies aiming to improve the targeting properties of PSs37 have reported the occurrence of interactions between anionic, cationic or non-charged Pcs with serum proteins.38,39 Only few reports, however, have also considered the fact that the excited states of PSs may be quenched following interactions with proteins.40,41 To study interactions of 1 with serum proteins, compound 1 was dissolved in serum-free culture medium (SFM) and then titrated with an increasing quantity of fetal bovine serum (FBS). The typical sharp shape of the absorption spectra of 1 did not change upon the addition of FBS, and only a small bathochromic shift of the Q-band from 654 nm to 661 nm was observed (Fig. 8a), indicating that the compound is transferred from water to a less polar environment and remains there in monomeric form. Fluorescence intensity substantially decreased upon the addition of FBS (Fig. 8b), with ΦF approaching approximately one-tenth of its original value at 10% FBS (Fig. 8c), a concentration that is typically used in serum-containing cell culture medium (SCM). These data suggest that binding of compound 1 to serum proteins substantially quenches its singlet excited state and makes it less active. The plot in Fig. 8c reached a plateau at 10% FBS in SFM, indicating that 1 is fully associated with serum proteins above this value. The emission maximum shifted from 657 nm to 665 nm concomitantly with the bathochromic shift of the absorption spectrum, indicating that the residual fluorescence originates from protein-associated 1 rather than from unbound molecules. For comparison, no interaction was detected for cationic Pc-Im16 during titration (Fig. 8c): its absorption spectrum and fluorescence remained unchanged upon addition of FBS.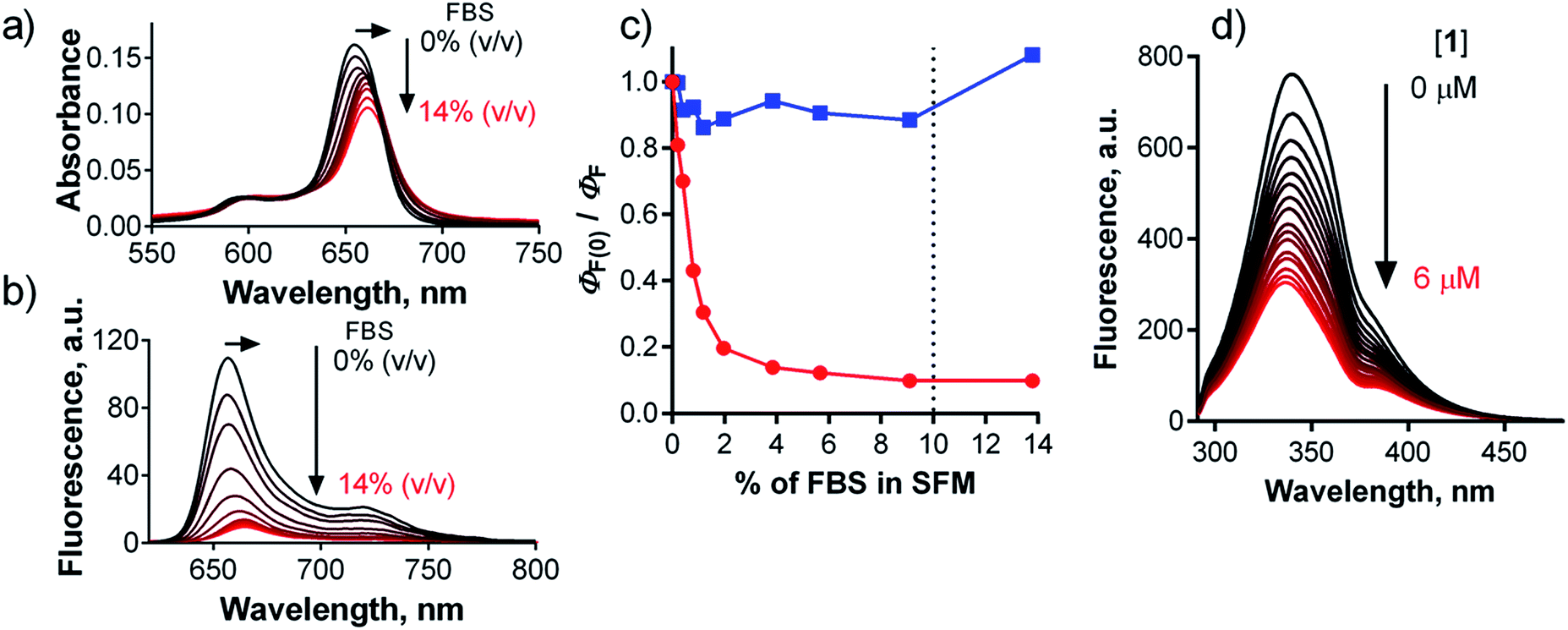 | ||
| Fig. 8 Changes in absorption (a) and emission (b) spectra of 1 (c = 1 μM) in SFM upon stepwise addition of FBS (up to 14% of volume). (c) Dependence of normalized ΦF of 1 (red) and Pc-Im16 (blue) in SFM on the concentration of FBS. The dotted line indicates the typical amount of FBS in SCM. (d) Changes of the fluorescence of BSA (c = 2 μM, λexc = 280 nm) in PBS upon the addition of a stock solution of 1 in PBS (c(1) = 0–6 μM). | ||
Based on the above results, the interaction of 1 with albumin, the most abundant protein in serum, was studied. Titration of 1 in SFM with increasing amounts of bovine serum albumin (BSA) gave similar results (shifts of the absorption and emission spectra, decreased ΦF) as those produced when 1 was titrated with FBS. The fluorescence signal decreased to approximately 40% of its original value at a typical BSA concentration in SCM (Fig. S34†). This can be explained by the fact that FBS contains approximately 38 mg mL−1 of total protein, of which albumin (although it is the most abundant) represents only 23 mg mL−1. In contrast, no interaction of Pc-Im16 with BSA was detected. Its fluorescence and absorption spectra remained unchanged upon addition of BSA, similar to its behavior in the experiment with FBS.
To quantify the interaction between BSA and 1, the binding constant (Kb) and number of binding sites (n) were determined. As BSA in phosphate-buffered saline (PBS) was titrated with compound 1, the fluorescence intensities of the BSA tryptophan residues were monitored at 340 nm; these values decreased (Fig. 8d). Based on the quenching data, the following values were obtained: Kb = 3.44 × 102 M−1, n = 0.56 (Fig. S34†). The Kb value is considerably lower (several orders of magnitude) than those reported in literature for the binding of proteins to anionic Pcs substituted with flexible peripheral substituents, indicating that the rigid position of the carboxylates in 1 significantly affects the strength of its interactions with proteins.39,40,42
These experiments revealed that compound 1 associates with BSA, interacting with tryptophan residues in the protein's pockets and leading to a mutual quenching of fluorescence. Additionally, a shielding effect from tryptophan residues that effectively quench singlet oxygens can be expected.43 These factors collectively contribute to the decreased photodynamic activity of 1 in cells. In contrast, the results obtained for Pc-Im16 indicate that rigid cationic substituents preclude any significant interactions with serum proteins, which is why they do not affect its photodynamic properties.
The effect of pH on the interactions between 1 and serum proteins was subsequently studied. Contrary to the results obtained in buffer (Fig. 6), the absorption spectra in SCM did not indicate aggregation at low pH but did show a bathochromic shift from 661 to 668 nm in response to a change of pH from 7.1 to 4.2 (Fig. 9a). The decrease in ΦF values was not as steep as in the buffer solution, although the original ΦF value of 0.04 at pH 7.1 in SCM was significantly decreased due to interactions between 1 and FBS proteins. The bathochromic shift of the absorption spectra indicates that the protonation of carboxylate functions of 1 at low pH leads to a tighter interaction of this compound with proteins. Therefore, the quenching of the fluorescence produced by 1 by tryptophan residues becomes more pronounced, leading to a decrease in ΦF. On the other hand, the interaction of 1 with proteins protects it from the self-aggregation that is induced at low pH in buffer solution. For this reason, significant fluorescence of 1 at pH 4.9 is still preserved in SCM, in contrast to the virtually complete deactivation of 1 in buffer (Fig. 9b, inset). These data are in good agreement with the results of the experiments with bafilomycin A1. The twofold enhancement in the fluorescence of 1 in cells upon treatment with this specific V-ATPase inhibitor (Fig. 7) corresponds well with the increase of ΦF from 0.012 to 0.027 when going from pH 4.9 to 6.0 in SCM. This also explains the relatively comparable fluorescence signals of 1 in endosomes and lysosomes, i.e., organelles with different pHs, and suggests that 1 is associated with serum proteins even intracellularly.
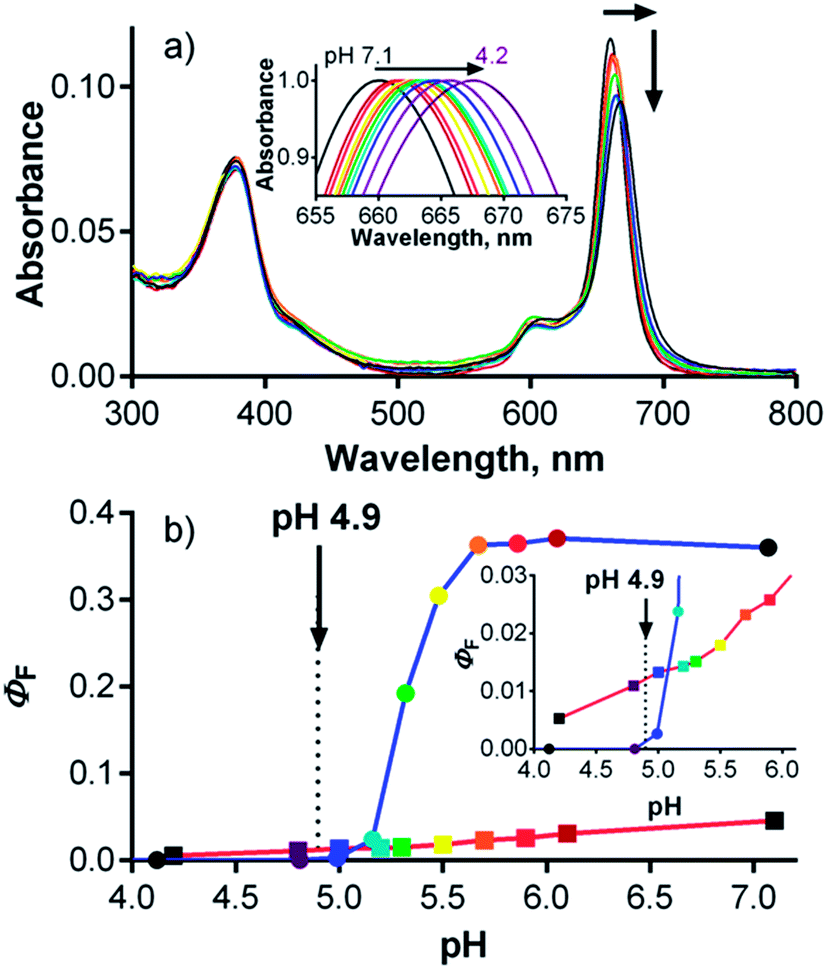 | ||
| Fig. 9 (a) Changes in the absorption spectra of 1 (c = 1 μM) in SCM at pHs ranging from 7.1 to 4.2. Inset: detail of normalized absorption spectra in Q-band region. (b) Dependence of ΦF on pH for 1 in SCM (red line, dots) and in buffer (blue line, squares) (λexc = 594 nm). Inset: enlarged part. The arrows indicate the lysosomal pH in HeLa cells as determined in this work. | ||
Further evidence for the significant influence of serum proteins on photodynamic activity was obtained by determining the phototoxicity of 1 dissolved in SFM on HeLa cells. A much lower IC50 value (0.55 ± 0.09 μM; Fig. S35†) was obtained due to the limited quenching of the excited states of 1 by proteins, which is fully concordant with the proposed concept.
Conclusions
In this study, a multistep protocol was developed to synthesize anionic TPyzPz bearing sixteen carboxylate functions on its periphery in a rigid arrangement. This arrangement proved to fully inhibit any aggregation of the planar hydrophobic cores of TPyzPzs in water or buffer at pH > 5.7. At low pH, protonation of the carboxylate functions led to a decrease in electrostatic repulsive forces and to substantial self-aggregation that made the compound inactive in buffer at pH < 4.8. Compound 1 exerted a modest photodynamic activity on HeLa cells in vitro with an IC50 values of 5.7 ± 1.1 μM and low dark toxicity (TC50 = 154 ± 8.5 μM). This represents a promising therapeutic ratio (TC50/IC50) of over 27. Although the IC50 value of 1 is comparable to those of similar anionic Pcs reported in literature, it is still one or two orders of magnitude higher than values generally reported for cationic Pcs. In addition to a slightly higher uptake of cationic Pc-Im16 in comparison to 1, several other factors appear to contribute to the significant differences between Pc-Im16 and compound 1. The slightly lower ΦΔ of 1 compared to Pc-Im16 seems to be of only marginal importance. Lysosomal pH was found to play a less significant role than originally suggested by the experiments in buffer, where the activity was expected to be completely eliminated below pH 4.8 by aggregation. In contrast to cationic Pc-Im16, which shows no interaction with serum proteins, tight binding of 1 to serum proteins led to an approximately tenfold drop in the photodynamic activity of 1 at pH 7.4. The association of 1 with proteins did, however, protect it from self-aggregation at low pH and preserved some photodynamic activity, even at pH < 4.8. Interestingly, published data suggest that protein–Pc associations may have a positive effect on the targeting of PS in vivo44 and may therefore ameliorate the negative impact of the quenching of the singlet excited state. Collectively, these experiments highlight important structure–activity relationships and provide insights that will aid the further development of anionic TPyzPz photosensitizers. Considering the published data on the photodynamic activities of anionic Pcs, this phenomenon is most likely present for other Pcs bearing carboxylate functions and should be considered in future investigations of similar PSs for PDT.Experimental section
General
All of the organic solvents used for synthesis were of analytical grade. The anhydrous butanol that was used for cyclotetramerization was freshly distilled from magnesium. All of the chemicals for synthesis and other anhydrous solvents were purchased from certified suppliers (Sigma Aldrich, TCI Europe, Acros, Merck) and used as received. Thin-layer chromatography was performed on Merck aluminum sheets coated with silica gel 60 F254. Merck Kieselgel 60 (0.040–0.063 mm) was used for column chromatography. Sephadex® G-25 (Aldrich) was used for gel filtration. Melting points were measured on an Electrothermal IA9200-series digital melting point apparatus (Electrothermal Engineering, Southend-on-Sea, Essex, UK). Infrared spectra were obtained on a Nicolet 6700 spectrometer using attenuated total reflectance technique. 1H and 13C NMR spectra were recorded on a Varian Mercury Vx BB 300 NMR spectrometer or a VNMR S500 NMR spectrometer. The chemical shifts are given relative to Si(CH3)4 and were locked to the signal of the solvent. J values are given in Hz. Elemental analyses were performed on an Automatic Microanalyzer EA1110CE (Fisons Instruments, Milan, Italy) or vario Micro Cube Elemental Analyzer (Elementar Analysensysteme GmbH, Hanau, Germany). UV-vis spectra were recorded using a Shimadzu UV-2600 spectrophotometer. Steady-state fluorescence spectra were measured using an AMINCO-Bowman Series 2 luminescence spectrometer. MALDI-TOF mass spectra were recorded in positive (2Mg, 2H, 2Zn) or negative (1) reflectron mode on a 4800 MALDI TOF/TOF mass spectrometer (AB Sciex, Framingham, MA, USA) in trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile, which was used as a matrix. The instrument was calibrated externally with a five-point calibration using a Peptide Calibration Mix1 kit (LaserBio Laboratories, Sophia-Antipolis, France). The purity of compound 1 was assessed by ion-pair chromatography and was found to be 97% (Fig. S22†). Separation was performed on a Hypersil BDS C18 column (100 × 4.6 mm, particle size 2.4 μm) using a mobile phase consisting of triethylamine acetate buffer (50 mM, pH 6.3, mobile phase A) and methanol (mobile phase B). A gradient elution was used. The gradient time program was set as follows: 0–20 min 20 → 40% B; 20–25 min 40% B; 25–30 min 40 → 85% B, which was held for 5 minutes. The column was then re-equilibrated for 5 min under the initial conditions. The column temperature was set at 40 °C, and the flow rate was set at 0.75 mL min−1. The compound was analyzed using a diode array detector, and chromatograms were recorded at wavelengths corresponding to the absorption maximum of the sample (647 nm). The chromatogram and peak similarity of the principal peak were assessed as the purity criteria. Compound Pc-Im16, which was previously published,7 was kindly supplied by Prof. Saad Makhseed (Kuwait University).Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800), 587 (20900), 370 (92600). νmax/cm−1 3396, 1612, 1559, 1420, 1362, 1256, 1194, 1121, 1091, 1011. δH (500 MHz; D2O; acetone) 8.51–8.48 (16H, m, ArH), 8.48–8.46 (8H, m, ArH). δC(125 MHz; D2O; acetone) 174.8, 155.9, 151.6, 149.1, 138.9, 137.6, 134.0, 130.7. m/z (MALDI-TOF) 1896.0 [M]−, 2009.0 [M + CF3COO]−, sample for MS was converted to free acid before spotting onto the MALDI plate.:10:1, 300 mL), and the suspension was stirred at rt for 30 min to remove unreacted magnesium butoxide. The green precipitate was collected by filtration and washed with water and methanol. The crude product was purified via column chromatography on silica with chloroform/THF (20:1) as the eluent. The pure product was dissolved in a minimum amount of chloroform and diluted with methanol. The fine green precipitate was collected and dried: dark green solid (840 mg, 59%). Found: C, 64.87; H, 6.79; N, 8.04. Calc. for C152H168MgN16O32·3H2O: C, 64.98; H, 6.24; N, 7.98. λmax (pyridine, 1 μM)/nm 663 (ε/dm3 mol−1 cm−1 287 900), 601 (33800), 373 nm (127100). νmax/cm−1 2962, 2873, 1724, 1603, 1541, 1459, 1386, 1279, 1240, 1141, 1091. δH(300 MHz; CDCl3/C5D5N 3:1; Me4Si) 9.0 (8H, t, J = 1.6, ArH), 8.94 (16H, d, J = 1.7, ArH), 4.47 (32H, t, J = 6.6, OCH2), 1.94–1.71 (32H, m, OCH2CH2), 1.65–1.45 (32H, m, CH2CH3), 1.07 (48H, t, CH3). δC (75 MHz; CDCl3/C5D5N 3:1; Me4Si) 165.3, 153.3, 151.3, 139.7, 136.0, 131.8, 131.6, 65.5, 30.9, 19.4, 13.9. m/z (MALDI-TOF) 2752.7 [M]+.:1) as the eluent. The pure product was washed with hexane and dried: dark green solid (561 mg, 81%). Found: C, 64.88; H, 6.45; N, 7.89. Calc. for C152H170N16O32·5H2O: C, 64.67; H, 6.45; N, 7.94. λmax (pyridine, 1 μM)/nm 666 (ε/dm3 mol−1 cm−1 200400), 607 (29500), 373 (100300). νmax/cm−1 3084, 2960, 2873, 1724, 1603, 1541, 1458, 1280, 1241, 1139, 1093. δH (300 MHz; CDCl3/C5D5N 3:1; Me4Si) 9.17–8.89 (m, ArH), 8.84 (d, J = 1.5, ArH), 8.78 (t, J = 1.5, ArH), 8.52–8.43 (m, ArH), 8.25–8.21 (m, ArH), 7.79 (br s, ArH), 4.78–4.02 (m, OCH2), 2.11–1.69 (m, OCH2CH2), 1.67–1.36 (m, CH2CH3), 1.18–0.81 (m, CH3) number of protons could not be assigned due to complexity of the spectra due to formation of supramolecular arrangement. δC (75 MHz; CDCl3/C5D5N 3:1; Me4Si) 165.43, 165.35, 165.18, 165.14, 164.96, 164.90, 164.83, 154.12, 154.03, 153.92, 140.21, 140.00, 139.11, 131.82, 131.50, 131.07, 110.41, 107.95, 67.33, 65.53, 65.41, 30.88, 30.85, 30.82, 30.71, 30.26, 29.59, 24.16, 19.40, 19.34, 19.28, 13.93, 13.90, 13.86, 13.81, 13.66. m/z (MALDI-TOF) 2731.1 [M]+.:1) as the eluent. The pure product was washed with hexane and dried: dark green solid (217 mg, 65%). Found: C, 63.21; H, 6.38; N, 7.81. Calc. for C152H168N16O32Zn·5H2O: C, 63.25; H, 6.22; N, 7.76. λmax (pyridine, 1 μM)/nm 659 (ε/dm3 mol−1 cm−1 306000), 596 (40200), 378 (156300). νmax/cm−1 3082, 2961, 2874, 1724, 1603, 1542, 1464, 1280, 1241, 1241, 1140, 1094. δH (300 MHz; CDCl3/C5D5N 3:1; Me4Si) 9.03–8.99 (8H, m, ArH), 8.98–8.94 (16H, m, ArH), 4.54–4.40 (32H, m, OCH2), 1.93–1.74 (32H, m, OCH2CH2), 1.61–1.43 (32H, m, CH2CH3), 1.17–0.99 (48H, m, CH3). δC(75 MHz; CDCl3/C5D5N 3:1; Me4Si) 165.3, 154.0, 152.0, 139.6, 136.1, 131.9, 131.7, 65.59, 30.9, 19.4, 13.9. m/z (MALDI-TOF) 2832.1 [M + K]+, 2816.1 [M + Na]+, 2793.1 [M]+.:1) as the eluent to recover starting aldehyde 6 (1.45 g, 12%) and then the pure product 7 was eluted: white crystals (9.0 g, 77%). Mp 127.3–127.6 °C. Found: C, 62.21; H, 5.38. Calc. for C26H26O10: C, 62.65; H, 5.26. νmax/cm−1 3088, 2988, 2941, 1729 (CO), 1682 (CO), 1599, 1445, 1371, 1323, 1242, 1184, 1106, 1024. δH (500 MHz, CDCl3, Me4Si) 8.96 (2H, t, J = 1.6, ArH), 8.81 (4H, d, J = 1.7, ArH), 4.44 (8H, q, J = 7.1, OCH2), 1.43 (12H, t, J = 7.1, CH3). δC(125 MHz, CDCl3, Me4Si) 190.9, 164.5, 136.2, 134.6, 133.2, 132.2, 62.0, 14.3.:1) as the eluent: yellow crystalline solid (1.75 g, 76%). Mp 145.4–145.7 °C. Found: C, 63.09; H, 5.16; N, 9.86. Calc. for C30H26N4O8: C, 63.15; H, 4.59; N, 9.82. νmax/cm−1 3089, 2988, 2910, 1719 (CO), 1600, 1365, 1314, 1246, 1212, 1140, 1097, 1029. δH (300 MHz, CDCl3, Me4Si) 8.75 (2H, t, J = 1.7, ArH), 8.41–8.30 (4H, m, ArH), 4.36 (8H, q, J = 7.2, OCH2), 1.35 (12H, t, J = 7.1, CH3). δC(75 MHz, CDCl3, Me4Si) 164.4, 153.6, 135.2, 134.5, 132.9, 132.0, 130.7, 112.6, 61.9, 14.2.:1, 100 mL) was added. The mixture was heated to 140 °C, and a light brown precipitate appeared after a couple of minutes. After 2 h of reaction, the mixture was cooled, acetic acid was removed under reduced pressure, and the mixture was stored in a fridge for 2 h. The solid was collected by filtration and washed with water to yield a light brown solid (3.28 g, 83%) of sufficient purity for the next reaction. The analytical sample was purified by crystallization from acetic acid, and light yellow crystals were obtained. Mp 373.5–374.5 °C (dec.). Found: C, 54.75; H, 3.04. Calc. for C18H10O10·½H2O: C, 54.70; H, 2.81. νmax/cm−1 1700, 1684, 1457, 1276, 1209, 1136. δH(300 MHz, CD3SOCD3, Me4Si) 13.72 (4H, br s, OH), 8.77–8.70 (6H, m, ArH). δC (75 MHz, CD3SOCD3, Me4Si) 189.7, 165.8, 135.2, 134.7, 133.5, 132.3.:3:1) as the eluent. The purified product was crystallized by slow addition of hexane into solution of 9 in acetone with few drops of methanol: white crystals (1.88 g, 47%). Mp 275–305 °C (dec.). Found: C, 54.46; H, 2.65; N, 11.46. Calc. for C22H10N4O8·1½H2O: C, 54.44; H, 2.70; N, 11.54. νmax/cm−1 1644, 1604, 1582, 1523, 1475, 1429, 1347, 1296, 1242. δH (300 MHz, CD3COCD3, Me4Si) 8.75–8.70 (2H, m, ArH), 8.44 (4H, d, J = 1.7, ArH). δC(75 MHz, CD3COCD3, Me4Si) 165.9, 155.2, 137.4, 135.8, 133.1, 132.7, 131.9, 114.5.
800), 587 (20900), 370 (92600). νmax/cm−1 3396, 1612, 1559, 1420, 1362, 1256, 1194, 1121, 1091, 1011. δH (500 MHz; D2O; acetone) 8.51–8.48 (16H, m, ArH), 8.48–8.46 (8H, m, ArH). δC(125 MHz; D2O; acetone) 174.8, 155.9, 151.6, 149.1, 138.9, 137.6, 134.0, 130.7. m/z (MALDI-TOF) 1896.0 [M]−, 2009.0 [M + CF3COO]−, sample for MS was converted to free acid before spotting onto the MALDI plate.:10:1, 300 mL), and the suspension was stirred at rt for 30 min to remove unreacted magnesium butoxide. The green precipitate was collected by filtration and washed with water and methanol. The crude product was purified via column chromatography on silica with chloroform/THF (20:1) as the eluent. The pure product was dissolved in a minimum amount of chloroform and diluted with methanol. The fine green precipitate was collected and dried: dark green solid (840 mg, 59%). Found: C, 64.87; H, 6.79; N, 8.04. Calc. for C152H168MgN16O32·3H2O: C, 64.98; H, 6.24; N, 7.98. λmax (pyridine, 1 μM)/nm 663 (ε/dm3 mol−1 cm−1 287 900), 601 (33800), 373 nm (127100). νmax/cm−1 2962, 2873, 1724, 1603, 1541, 1459, 1386, 1279, 1240, 1141, 1091. δH(300 MHz; CDCl3/C5D5N 3:1; Me4Si) 9.0 (8H, t, J = 1.6, ArH), 8.94 (16H, d, J = 1.7, ArH), 4.47 (32H, t, J = 6.6, OCH2), 1.94–1.71 (32H, m, OCH2CH2), 1.65–1.45 (32H, m, CH2CH3), 1.07 (48H, t, CH3). δC (75 MHz; CDCl3/C5D5N 3:1; Me4Si) 165.3, 153.3, 151.3, 139.7, 136.0, 131.8, 131.6, 65.5, 30.9, 19.4, 13.9. m/z (MALDI-TOF) 2752.7 [M]+.:1) as the eluent. The pure product was washed with hexane and dried: dark green solid (561 mg, 81%). Found: C, 64.88; H, 6.45; N, 7.89. Calc. for C152H170N16O32·5H2O: C, 64.67; H, 6.45; N, 7.94. λmax (pyridine, 1 μM)/nm 666 (ε/dm3 mol−1 cm−1 200400), 607 (29500), 373 (100300). νmax/cm−1 3084, 2960, 2873, 1724, 1603, 1541, 1458, 1280, 1241, 1139, 1093. δH (300 MHz; CDCl3/C5D5N 3:1; Me4Si) 9.17–8.89 (m, ArH), 8.84 (d, J = 1.5, ArH), 8.78 (t, J = 1.5, ArH), 8.52–8.43 (m, ArH), 8.25–8.21 (m, ArH), 7.79 (br s, ArH), 4.78–4.02 (m, OCH2), 2.11–1.69 (m, OCH2CH2), 1.67–1.36 (m, CH2CH3), 1.18–0.81 (m, CH3) number of protons could not be assigned due to complexity of the spectra due to formation of supramolecular arrangement. δC (75 MHz; CDCl3/C5D5N 3:1; Me4Si) 165.43, 165.35, 165.18, 165.14, 164.96, 164.90, 164.83, 154.12, 154.03, 153.92, 140.21, 140.00, 139.11, 131.82, 131.50, 131.07, 110.41, 107.95, 67.33, 65.53, 65.41, 30.88, 30.85, 30.82, 30.71, 30.26, 29.59, 24.16, 19.40, 19.34, 19.28, 13.93, 13.90, 13.86, 13.81, 13.66. m/z (MALDI-TOF) 2731.1 [M]+.:1) as the eluent. The pure product was washed with hexane and dried: dark green solid (217 mg, 65%). Found: C, 63.21; H, 6.38; N, 7.81. Calc. for C152H168N16O32Zn·5H2O: C, 63.25; H, 6.22; N, 7.76. λmax (pyridine, 1 μM)/nm 659 (ε/dm3 mol−1 cm−1 306000), 596 (40200), 378 (156300). νmax/cm−1 3082, 2961, 2874, 1724, 1603, 1542, 1464, 1280, 1241, 1241, 1140, 1094. δH (300 MHz; CDCl3/C5D5N 3:1; Me4Si) 9.03–8.99 (8H, m, ArH), 8.98–8.94 (16H, m, ArH), 4.54–4.40 (32H, m, OCH2), 1.93–1.74 (32H, m, OCH2CH2), 1.61–1.43 (32H, m, CH2CH3), 1.17–0.99 (48H, m, CH3). δC(75 MHz; CDCl3/C5D5N 3:1; Me4Si) 165.3, 154.0, 152.0, 139.6, 136.1, 131.9, 131.7, 65.59, 30.9, 19.4, 13.9. m/z (MALDI-TOF) 2832.1 [M + K]+, 2816.1 [M + Na]+, 2793.1 [M]+.:1) as the eluent to recover starting aldehyde 6 (1.45 g, 12%) and then the pure product 7 was eluted: white crystals (9.0 g, 77%). Mp 127.3–127.6 °C. Found: C, 62.21; H, 5.38. Calc. for C26H26O10: C, 62.65; H, 5.26. νmax/cm−1 3088, 2988, 2941, 1729 (CO), 1682 (CO), 1599, 1445, 1371, 1323, 1242, 1184, 1106, 1024. δH (500 MHz, CDCl3, Me4Si) 8.96 (2H, t, J = 1.6, ArH), 8.81 (4H, d, J = 1.7, ArH), 4.44 (8H, q, J = 7.1, OCH2), 1.43 (12H, t, J = 7.1, CH3). δC(125 MHz, CDCl3, Me4Si) 190.9, 164.5, 136.2, 134.6, 133.2, 132.2, 62.0, 14.3.:1) as the eluent: yellow crystalline solid (1.75 g, 76%). Mp 145.4–145.7 °C. Found: C, 63.09; H, 5.16; N, 9.86. Calc. for C30H26N4O8: C, 63.15; H, 4.59; N, 9.82. νmax/cm−1 3089, 2988, 2910, 1719 (CO), 1600, 1365, 1314, 1246, 1212, 1140, 1097, 1029. δH (300 MHz, CDCl3, Me4Si) 8.75 (2H, t, J = 1.7, ArH), 8.41–8.30 (4H, m, ArH), 4.36 (8H, q, J = 7.2, OCH2), 1.35 (12H, t, J = 7.1, CH3). δC(75 MHz, CDCl3, Me4Si) 164.4, 153.6, 135.2, 134.5, 132.9, 132.0, 130.7, 112.6, 61.9, 14.2.:1, 100 mL) was added. The mixture was heated to 140 °C, and a light brown precipitate appeared after a couple of minutes. After 2 h of reaction, the mixture was cooled, acetic acid was removed under reduced pressure, and the mixture was stored in a fridge for 2 h. The solid was collected by filtration and washed with water to yield a light brown solid (3.28 g, 83%) of sufficient purity for the next reaction. The analytical sample was purified by crystallization from acetic acid, and light yellow crystals were obtained. Mp 373.5–374.5 °C (dec.). Found: C, 54.75; H, 3.04. Calc. for C18H10O10·½H2O: C, 54.70; H, 2.81. νmax/cm−1 1700, 1684, 1457, 1276, 1209, 1136. δH(300 MHz, CD3SOCD3, Me4Si) 13.72 (4H, br s, OH), 8.77–8.70 (6H, m, ArH). δC (75 MHz, CD3SOCD3, Me4Si) 189.7, 165.8, 135.2, 134.7, 133.5, 132.3.:3:1) as the eluent. The purified product was crystallized by slow addition of hexane into solution of 9 in acetone with few drops of methanol: white crystals (1.88 g, 47%). Mp 275–305 °C (dec.). Found: C, 54.46; H, 2.65; N, 11.46. Calc. for C22H10N4O8·1½H2O: C, 54.44; H, 2.70; N, 11.54. νmax/cm−1 1644, 1604, 1582, 1523, 1475, 1429, 1347, 1296, 1242. δH (300 MHz, CD3COCD3, Me4Si) 8.75–8.70 (2H, m, ArH), 8.44 (4H, d, J = 1.7, ArH). δC(75 MHz, CD3COCD3, Me4Si) 165.9, 155.2, 137.4, 135.8, 133.1, 132.7, 131.9, 114.5.Photophysical measurements
The singlet oxygen quantum yields (ΦΔ) and the fluorescence quantum yields (ΦF) of 2Mg, 2H and 2Zn in pyridine and ΦF of 1 in water or phosphate buffer (pH 7.1) were determined using published comparative methods with unsubstituted ZnPc (obtained from Sigma-Aldrich) as a reference compound.44 The following values for ZnPc were used in the calculations: ΦΔ(pyridine) = 0.61,45 and ΦF(THF) = 0.32.28 The emission spectra were obtained after excitation of the sample and the reference at λexc = 599 nm and were corrected for instrument response. All of the determinations were performed in triplicate, and the data represent the means of these measurements. The estimated experimental error was ±10%.For sample 1 in D2O (or in D2O 0.1 M phosphate buffer, pD = 7.1), the near-infrared luminescence of singlet oxygen O2(1Δg) at 1270 nm was monitored using a Judson Ge diode and interference filter. The samples were excited with a Lambda Physik FL3002 dye laser (λexc = 650 nm). The signal from the detector was collected in a 600 MHz oscilloscope (Agilent Infiniium) and transferred to a computer for further analysis. The initial part (up to 10 μs) failed due to light scattering and strong fluorescence and was not used for evaluation. The signal-to-noise ratio of the signals was improved by averaging 500 individual traces. The quantum yield of singlet oxygen, ΦΔ, was estimated by a comparative method using methylene blue as a reference (ΦΔ = 0.52 in D2O).46 Incident energy used was within the energy region where the intensity of a luminescence signal was directly proportional to the incident energy (less than 2 mJ). To see the original data in graphical form, refer to the ESI (Fig. S23 and S24†).
Spectral and photophysical data for 1 at different pHs were obtained using phosphate–citrate buffer (0.2 M sodium hydrogen phosphate and 0.1 M citric acid). pH was determined using a Hanna pH209 pH meter that was calibrated with a two-point calibration at pH 4.00 and 7.00 before measurements. The fluorescence was monitored after excitation at 594 nm.
Cell cultures
A human cervical carcinoma cell line (HeLa) was purchased from the American Type Cell Culture Collection (USA). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) without phenol red (Lonza, Belgium). The medium was supplemented with 10% heat-inactivated fetal bovine serum (FBS; Sigma, Germany), 1% penicillin/streptomycin solution (Lonza), 10 mM HEPES buffer (Sigma) and 4 mM L-glutamine (Lonza). For the purposes of this study, serum-free medium (SFM) was also used in some experiments. SFM contained DMEM with all additives except FBS. The cells were incubated in 75 cm2 tissue culture flasks (TPP, Switzerland), maintained in an incubator at 37 °C in a humidified atmosphere of 5% CO2 and subcultured every 3–4 days. For the cytotoxicity experiments (phototoxicity and dark toxicity), the cells were seeded in 96-well plates (TPP) at a density of 7.5 × 103 cells per well for 24 h. A stock solution of compound 1 was prepared in serum-containing medium (SCM) at a concentration of 500 μM and sterilized by filtration through a 0.22 μm syringe filter. The concentration of filtered compound 1 was verified by absorbance measurements. The absorbance remained the same before and after filtration.Cellular uptake
To establish a time profile of intracellular accumulation, HeLa cells were seeded into 6 cm Petri dishes (TPP) at a density of 4.5 × 104 cells per dish. The cells were left to attach for 24 h, the medium was removed, and 4 μM compound 1 or Pc-Im16 in 5 mL of cultivation medium was added. The cells were washed three times with 5 mL phosphate-buffered saline (PBS, Sigma) after a selected time, followed by the addition of 5 mL of PBS. The cells were scraped, transferred to 15 mL flasks and centrifuged. The supernatant was completely removed, and 500 μL of water was added. Cell lysis was performed with two freeze (−80 °C) thaw (37 °C) cycles over 24 h. The fluorescence intensities of 1 (λem = 663 nm, λexc = 375 nm, addition of 10 μL of 1 M NaOH) and Pc-Im16 (λem = 691 nm, λexc = 363 nm) were measured after and plotted against the incubation time. The uptake experiments were performed in duplicates. Calibration curves were constructed using dilutions of PS stock solutions in SCM with the cell lysate prepared as described above. The total amount of protein in each sample was determined using a bicinchoninic acid assay described by Smith et al.47 with 10 μL of cell lysate (performed in triplicate). Optical density (λ = 562 nm) was measured after a 30 min incubation at 37 °C (Tecan Infinite 200 M plate reader). A calibration curve for protein determination was constructed using various concentrations of BSA in milliQ water (50–1500 μg mL−1).Subcellular localization
HeLa cells were seeded on Petri dishes suitable for confocal microscopy (WillCo Wells, The Netherlands) at a density of 6 × 104 cells per dish in SCM. For detection of mitochondria and lysosomes, the cells were incubated for 12 h with 10 μM 1 at 37 °C under a 5% CO2 atmosphere and constant humidity. The medium was removed, the cells were washed twice with pre-warmed PBS, and fresh medium was added. The cells were incubated with LysoTracker Blue DND-22 (Molecular Probes, 1 μM) and MitoTracker Green FM (Molecular Probes, 1 μM) for an additional 10 min. For detection of endocytic pathways, the cells were co-incubated with 10 μM 1 and dextran-FITC (3000 MW; Molecular Probes, 0.5 mg mL−1) for 5 h. After the incubation, the cells were rinsed twice with pre-warmed PBS and examined under a Nikon Eclipse Ti (Nikon, Japan) fluorescence microscope equipped with an Andor Zyla 5.5 cooled digital sCMOS camera (Andor Technology, UK) and NIS Elements AR 4.20 software (Laboratory Imaging, Czech Republic).Lamp1-RFP and Rab7a-GFP transfection
To visualize endo-lysosomal vesicles, we transfected cells using the baculovirus-based expression vectors CellLight® Late Endosomes-GFP, BacMam 2.0 (Molecular Probes) and CellLight® Lysosomes-RFP, BacMam 2.0 (Molecular Probes) to express fluorescent Rab7a and Lamp1 proteins, respectively. HeLa cells were seeded on Petri dishes suitable for confocal microscopy (WillCo Wells) at a density of 4 × 104 cells per dish. Once they attached to the surface, both reagents were added at a concentration of 50 particles per cell. Compound 1 (4 μM) was added 12 h before the end of the incubation. After 24 h, the cells were stained with Hoechst 33342 (5 nM, Molecular Probes) for 15 min, rinsed twice with pre-warmed PBS and 2 mL of pre-warmed ADS buffer (116.0 mM NaCl, 5.3 mM KCl, 1.2 mM MgSO4·7H2O, 1.13 mM NaH2PO5·2H2O, 20 mM HEPES in MQ-H2O) with 1 mM CaCl2 and 5 mM glucose was added. Microphotographs of cells successfully transfected with both vectors were obtained using a Nikon Eclipse Ti fluorescence microscope equipped with an Andor Zyla 5.5 sCMOS monochromatic camera.Cytotoxicity and photodynamic treatment
Dark toxicity (inherent toxicity of compound 1 without the presence of light) was assessed over a wide concentration range (0.1–500 μM) after a 24 h incubation in HeLa cells. Viabilities were determined using a Neutral Red (NR) uptake assay (Sigma). The absorbance of soluble NR at λ = 540 nm was measured using a Tecan Infinite 200 M plate reader. The viability of each experimental group was expressed as a percentage of the untreated controls incubated under the same conditions.For the photodynamic treatment experiments, HeLa cells were first incubated with various concentrations of 1 (0.01–50 μM) for 12 h. The cells were than washed, fresh medium was added, and the cells were irradiated for 15 min using a 450 W ozone-free Xe lamp (Newport) with intensity reduced to 400 W (λ > 570 nm, 12.4 mW cm−2, 11.2 J cm−2) that was equipped with a long-pass filter (Newport OG570) and a water filter (8 cm) to eliminate undesirable wavelengths and heat radiation. Cellular viability was measured after an additional 24 h incubation by NR as previously described. At least four independent experiments, each in quadruplicate, were performed. The concentration of 1 that induced a 50% viability decrease after treatment under dark conditions (TC50 is the median toxic concentration) or after photodynamic treatment (IC50 is the median inhibition concentration) were calculated using GraphPad Prism 6.04 software (GraphPad Software, Inc. San Diego, CA) for each independent experiment. The data are presented as the means (±standard deviation) of these values.
Lysosomal pH measurements
For lysosomal ratiometric pH measurement, cells were seeded in black 96-well plates with transparent bottoms (Corning, USA) at a density of 7.5 × 103 cells per well. The cells were then washed twice with SFM, and fresh medium containing LysoSensor Yellow/Blue DND-160 (Molecular Probes, 2 μM) was added for 5 min. After incubation, the cells were washed twice with PBS, and fresh medium was added. Fluorescence measurements with and without LysoSensor Yellow/Blue DND-160 were performed at 37 °C using a Tecan Infinite M 200 plate reader (excitation λ = 329, 384 nm and emission λ = 440, 540 nm). A series of standard solutions (SFM; pH = 2.5–8.5) was prepared at 37 °C. LysoSensor Yellow/Blue DND-160 (Molecular Probes, 10 μM) was added, and the fluorescence was measured using a Tecan Infinite M 200 as described above. A calibration curve was prepared with the fluorescence ratio dependence on pH (Fig. S32†).Bafilomycin A1 inhibition of V-ATPase
HeLa cells were seeded in black 96-well plates with transparent bottoms (Corning) at a density of 7.5 × 103 cells per well and incubated with 4 μM of 1 or Pc-Im16 for 12 h. Bafilomycin A1 (Sigma) at a final concentration of 1 μM was added 3, 2, 1 and 0.5 h prior to fluorescence measurement. The cells were washed twice with pre-warmed PBS, and fresh medium was added. Fluorescence (λexc = 375 nm, λem = 671 nm for 1 and λexc = 363 nm, λem = 693 nm for Pc-Im16) was measured using a Tecan Infinite M 200. The results were expressed relative to a control treated under the same conditions with 1 or Pc-Im16 but in the absence of bafilomycin A1. Two independent experiments, each with ten replicates, were performed.Interactions with BSA and serum proteins
Solutions of compound 1 or Pc-Im16 (c = 1 μM) in SFM were titrated with FBS up to at least 10% (v/v) in SFM, which is the typical concentration of serum in cell culture medium. Fluorescence was monitored after excitation at 594 nm. The same experiment was performed with BSA instead of FBS.To determine the binding constant (Kb), 2.5 mL of BSA solution (c = 2 μM) in PBS was titrated with 1 (c = 500 μM) in PBS, and the fluorescence of tryptophan residues was monitored at 340 nm after excitation at 280 nm. The binding constant (Kb) and number of binding sites (n) were determined using eqn (1) with the corresponding plot of the data on a double logarithmic scale (Fig. S34†):
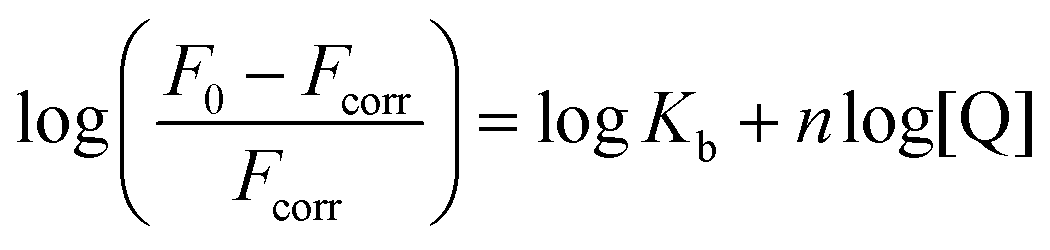 | (1) |
| Fcorr = Fobs10(Aex+Aem)/2 | (2) |
Data analysis
Statistical analysis was performed using GraphPad Prism. One-way ANOVA with Bonferroni's multiple comparisons post hoc test was used. The results were compared with those produced by the control samples, and the means were considered significant if (*) p < 0.05, (**) p < 0.01, and (***) p < 0.001.Acknowledgements
This work was supported by the Czech Science Foundation (No. 13-27761S) and Charles University in Prague (SVV 260 183). The authors would like to thank Jiří Kuneš for the NMR measurements and Juraj Lenčo for the MALDI-TOF MS measurements. The kind donation of Pc-Im16 from Prof. Saad Makhseed (Kuwait University) is also acknowledged.Notes and references
- D. Wöhrle, G. Schnurpfeil, S. G. Makarov, A. Kazarin and O. N. Suvorova, Macroheterocycles, 2012, 5, 191–202 CrossRef.
- M. Machacek, A. Cidlina, V. Novakova, J. Svec, E. Rudolf, M. Miletin, R. Kučera, T. Simunek and P. Zimcik, J. Med. Chem., 2015, 58, 1736–1749 CrossRef CAS PubMed.
- D. K. P. Ng, Future Med. Chem., 2014, 6, 1991–1993 CrossRef CAS PubMed; B. G. Ongarora, Z. Zhou, E. A. Okoth, I. Kolesnichenko, K. M. Smith and M. G. H. Vicente, J. Porphyrins Phthalocyanines, 2014, 18, 1021–1033 CrossRef PubMed.
- S. Singh, A. Aggarwal, N. V. Bhupathiraju, G. Arianna, K. Tiwari and C. M. Drain, Chem. Rev., 2015, 115, 10261–10306 CrossRef CAS PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- F. Dumoulin, M. Durmus, V. Ahsen and T. Nyokong, Coord. Chem. Rev., 2010, 254, 2792–2847 CrossRef CAS.
- S. Makhseed, M. Machacek, W. Alfadly, A. Tuhl, M. Vinodh, T. Simunek, V. Novakova, P. Kubat, E. Rudolf and P. Zimcik, Chem. Commun., 2013, 49, 11149–11151 RSC.
- X.-S. Li, J. Guo, J.-J. Zhuang, B.-Y. Zheng, M.-R. Ke and J.-D. Huang, Bioorg. Med. Chem. Lett., 2015, 25, 2386–2389 CrossRef CAS PubMed.
- W. Liu, T. J. Jensen, F. R. Fronczek, R. P. Hammer, K. M. Smith and M. G. H. Vicente, J. Med. Chem., 2005, 48, 1033–1041 CrossRef CAS PubMed.
- J. T. F. Lau, P.-C. Lo, W.-P. Fong and D. K. P. Ng, Chem.–Eur. J., 2011, 17, 7569–7577 CrossRef CAS PubMed; H. Li, T. J. Jensen, F. R. Fronczek and M. G. Vicente, J. Med. Chem., 2008, 51, 502–511 CrossRef PubMed.
- M. Nishida, H. Horiuchi, A. Momotake, Y. Nishimura, H. Hiratsuka and T. Arai, J. Porphyrins Phthalocyanines, 2011, 15, 47–53 CrossRef CAS.
- N. A. Kuznetsova, N. S. Gretsova, V. M. Derkacheva, S. A. Mikhalenko, L. I. Solov'eva, O. A. Yuzhakova, O. L. Kaliya and E. A. Luk'yanets, Russ. J. Gen. Chem., 2002, 72, 300–306 CrossRef CAS; O. G. Lutsenko, V. P. Kulinich, G. P. Shaposhnikov and A. V. Lyubomtsev, Russ. J. Gen. Chem., 2004, 74, 286–291 CrossRef; W. M. Sharman, S. V. Kudrevich and J. E. van Lier, Tetrahedron Lett., 1996, 37, 5831–5834 CrossRef.
- M. P. Donzello, C. Ercolani, V. Novakova, P. Zimcik and P. A. Stuzhin, Coord. Chem. Rev., 2016, 309, 107–179 CrossRef CAS.
- V. Novakova, L. Lochman, I. Zajícová, K. Kopecky, M. Miletin, K. Lang, K. Kirakci and P. Zimcik, Chem.–Eur. J., 2013, 19, 5025–5028 CrossRef CAS PubMed; L. Lochman, J. Svec, J. Roh and V. Novakova, Dyes Pigm., 2015, 121, 178–187 CrossRef.
- K. Kopecky, V. Novakova, M. Miletin, R. Kucera and P. Zimcik, Bioconjugate Chem., 2010, 21, 1872–1879 CrossRef CAS PubMed.
- V. Novakova, P. Reimerova, J. Svec, D. Suchan, M. Miletin, H. M. Rhoda, V. N. Nemykin and P. Zimcik, Dalton Trans., 2015, 44, 13220–13233 RSC.
- P. Zimcik, M. Miletin, H. Radilova, V. Novakova, K. Kopecky, J. Svec and E. Rudolf, Photochem. Photobiol., 2010, 86, 168–175 CrossRef CAS PubMed.
- P. Zimcik, V. Novakova, M. Miletin and K. Kopecky, Macroheterocycles, 2008, 1, 21–29 CAS.
- M. Kathiresan, L. Walder, F. Ye and H. Reuter, Tetrahedron Lett., 2010, 51, 2188–2192 CrossRef CAS.
- J. W. Leon, M. Kawa and J. M. J. Frechet, J. Am. Chem. Soc., 1996, 118, 8847–8859 CrossRef CAS.
- J. T. Dy, K. Ogawa, A. Satake, A. Ishizumi and Y. Kobuke, Chem.–Eur. J., 2007, 13, 3491–3500 CrossRef CAS PubMed.
- A. Tuhl, S. Makhseed, P. Zimcik, N. Al-Awadi, V. Novakova and J. Samuel, J. Porphyrins Phthalocyanines, 2012, 16, 817–825 CrossRef CAS.
- V. Novakova, P. Zimcik, K. Kopecky, M. Miletin, J. Kuneš and K. Lang, Eur. J. Org. Chem., 2008, 2008, 3260–3263 CrossRef.
- C. F. Choi, P. T. Tsang, J. D. Huang, E. Y. M. Chan, W. H. Ko, W. P. Fong and D. K. P. Ng, Chem. Commun., 2004, 2236–2237 RSC.
- S. V. Kudrevich, M. G. Galpern and J. E. van Lier, Synthesis, 1994, 779–781 CrossRef CAS.
- P. A. Stuzhin and O. G. Khelevina, Coord. Chem. Rev., 1996, 147, 41–86 CrossRef CAS.
- X. Y. Li, X. He, A. C. H. Ng, C. Wu and D. K. P. Ng, Macromolecules, 2000, 33, 2119–2123 CrossRef CAS; F. Setaro, R. Ruiz-González, S. Nonell, U. Hahn and T. Torres, J. Inorg. Biochem., 2014, 136, 170–176 CrossRef PubMed.
- P. Zimcik, V. Novakova, K. Kopecky, M. Miletin, R. Z. Uslu Kobak, E. Svandrlikova, L. Váchová and K. Lang, Inorg. Chem., 2012, 51, 4215–4223 CrossRef CAS PubMed.
- M. K. Johansson and R. M. Cook, Chem.–Eur. J., 2003, 9, 3466–3471 CrossRef CAS PubMed.
- I. Mellman, R. Fuchs and A. Helenius, Annu. Rev. Biochem., 1986, 55, 663–700 CrossRef CAS PubMed.
- P. Saftig and J. Klumperman, Nat. Rev. Mol. Cell Biol., 2009, 10, 623–635 CrossRef CAS PubMed.
- P. A. Vanlandingham and B. P. Ceresa, J. Biol. Chem., 2009, 284, 12110–12124 CrossRef CAS PubMed.
- S. Meresse, J. P. Gorvel and P. Chavrier, J. Cell Sci., 1995, 108, 3349–3358 Search PubMed; C. Bucci, P. Thomsen, P. Nicoziani, J. McCarthy and B. van Deurs, Mol. Biol. Cell, 2000, 11, 467–480 CrossRef CAS PubMed.
- W. H. Humphries, C. J. Szymanski and C. K. Payne, PLoS One, 2011, 6, e26626 CAS.
- T. Yoshimori, A. Yamamoto, Y. Moriyama, M. Futai and Y. Tashiro, J. Biol. Chem., 1991, 266, 17707–17712 CAS.
- B. Poole and S. Ohkuma, J. Cell Biol., 1981, 90, 665–669 CrossRef CAS PubMed.
- W. M. Sharman, J. E. van Lier and C. M. Allen, Adv. Drug Delivery Rev., 2004, 56, 53–76 CrossRef CAS PubMed; D. Xu, X. Chen, K. e. Chen, Y. Peng, Y. Li, Y. Ke and D. Gan, J. Biomater. Appl., 2014, 29, 378–385 CrossRef PubMed.
- S. Alpugan, G. Garcia, F. Poyer, M. Durmus, P. Maillard, V. Ahsen and F. Dumoulin, J. Porphyrins Phthalocyanines, 2013, 17, 596–603 CrossRef CAS; J.-D. Huang, P.-C. Lo, Y.-M. Chen, J. C. Lai, W.-P. Fong and D. K. P. Ng, J. Inorg. Biochem., 2006, 100, 946–951 CrossRef PubMed.
- H.-N. Xu, H.-J. Chen, B.-Y. Zheng, Y.-Q. Zheng, M.-R. Ke and J.-D. Huang, Ultrason. Sonochem., 2015, 22, 125–131 CrossRef CAS PubMed.
- A. Ogunsipe and T. Nyokong, Photochem. Photobiol. Sci., 2005, 4, 510–516 CAS.
- K. Lang, D. M. Wagnerova, P. Engst and P. Kubat, Z. Phys. Chem., 1994, 187, 213–221 CrossRef CAS.
- N. S. Lebedeva, T. E. Popova, E. A. Mal'kova and Y. A. Gubarev, Russ. J. Phys. Chem. A, 2013, 87, 2030–2033 CrossRef CAS.
- K. Hirakawa, H. Umemoto, R. Kikuchi, H. Yamaguchi, Y. Nishimura, T. Arai, S. Okazaki and H. Segawa, Chem. Res. Toxicol., 2015, 28, 262–267 CAS.
- V. Novakova, M. Miletin, T. Filandrová, J. Lenčo, A. Růžička and P. Zimcik, J. Org. Chem., 2014, 79, 2082–2093 CrossRef CAS PubMed.
- A. Ogunsipe, D. Maree and T. Nyokong, J. Mol. Struct., 2003, 650, 131–140 CrossRef CAS.
- K. K. Chin, C. C. Trevithick-Sutton, J. McCallum, S. Jockusch, N. J. Turro, J. C. Scaiano, C. S. Foote and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2008, 130, 6912–6913 CrossRef CAS PubMed.
- P. K. Smith, R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson and D. C. Klenk, Anal. Biochem., 1985, 150, 76–85 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of known compounds, NMR and mass spectra, chromatograms, absorption and fluorescence spectra, photophysical characterization, table with photodynamic activity of known Pcs, additional photos from fluorescence microscopy. See DOI: 10.1039/c5ra25881b |
| This journal is © The Royal Society of Chemistry 2016 |