
Retracted Article: CuS nanocrystal@microgel nanocomposites for light-regulated release of dual-drugs and chemo-photothermal synergistic therapy in vitro†
Abstract
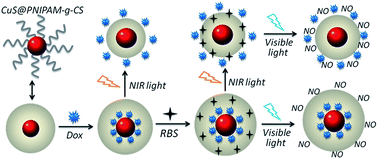
In this article, we described the synthesis of novel light-sensitive inorganic@organic core/shell nanocomposites that consisted of CuS nanocrystals as the core and a poly(N-isopropylacrylamide)-graft-chitosan (PNIPAM-g-CS) microgel as the shell. The CuS@PNIPAM-g-CS nanocomposites were synthesized by temperature-tunable copolymerization of NIPAM and CS in the presence of CuS nanocrystals (∼5.4 nm). The nanocomposites showed an average diameter of ∼56 nm and a strong longitudinal surface plasmon band in the near-infrared (NIR) region. Due to the photothermal effect of CuS under NIR light (980 nm) irradiation, the nanocomposites presented photothermal-sensitive volume shrinkage of the PNIPAM-g-CS microgel. After loading of doxorubicin (Dox), the nanocomposites were utilized as versatile nanocarriers for photothermal-induced release of Dox. After loading of Dox, nitric oxide (NO) photodonors (RBS) were then loaded into nanocomposites to fabricate Dox/RBS dual-loading CuS@PNIPAM-g-CS nanocarriers. Upon visible light (365 nm) irradiation, the nanocarriers could release NO due to the photolysis of RBS. Experimental results implied that NIR and visible light, respectively, triggered the release of Dox and NO from the nanocarriers. Together with the photothermal effect of CuS, the nanocarriers simultaneously realized the light-triggered release of dual-drugs and synergistic chem-photothermal therapy to cancer cells in vitro.
 Please wait while we load your content...
Please wait while we load your content...

