
Giant enhancement of upconversion emission in NaYF4:Er3+@NaYF4:Yb3+ active-core/active-shell nanoparticles
Abstract
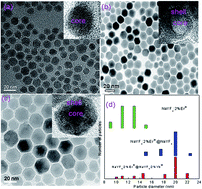
Varying the type and the doping concentration of rare-earth ions was reported as a conventional method to tune upconversion emissions of rare-earth ions doped nanoparticles (NPs). In this work, an effective method to enhance greatly the infrared-visible upconversion emissions of Er3+ ions is demonstrated through coating an optimized active NaYF4:Yb3+ shell around NaYF4:Er3+ core nanoparticles. Phase, shape, and size of the resulting core–shell NPs are studied by X-ray diffraction and high-resolution transmission electron microscopy. Compared with the core-only NaYF4:Er3+ NPs, a maximum 97.56-fold overall enhancement in the emission intensity of Er3+ ions in the core–shell NPs is achieved under 980 nm infrared excitation, and a maximum 38.6-fold overall enhancement is achieved under 1540 nm infrared excitation. The luminescence enhancement effect and color output exhibits a strong dependence on the doping concentrations of Yb3+ in the NaYF4:Yb3+ active-shell. By analyzing the decay lifetimes of the emission bands, the giant luminescence enhancement is due to the significant increase in the near-infrared absorption and efficient energy transfer from Yb3+ primary-sensitizers to Er3+ activators via Yb3+ bridging sensitizers.
 Please wait while we load your content...
Please wait while we load your content...

