
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure and biological activities of metal complexes of flumequine†
Ifigenia
Tsitsa
a,
Alketa
Tarushi
a,
Panagiota
Doukoume
b,
Franc
Perdih
c,
Andreia
de Almeida
d,
Athanasios
Papadopoulos
b,
Stavros
Kalogiannis
b,
Angela
Casini
*de,
Iztok
Turel
*c and
George
Psomas
*a
aDepartment of General and Inorganic Chemistry, Faculty of Chemistry, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece. E-mail: gepsomas@chem.auth.gr; Fax: +30 2310997738; Tel: +30 2310997790
bDepartment of Nutrition and Dietetics, Faculty of Agriculture, Food Technology and Nutrition, Alexander Technological Educational Institution, Sindos, Thessaloniki, Greece
cFaculty of Chemistry and Chemical Technology, University of Ljubljana, Vecna pot 113, 1000 Ljubljana, Slovenia. E-mail: iztok.turel@fkkt.uni-lj.si; Fax: +386-1-2419144; Tel: +386-1-4798525
dDepartment of Pharmacokinetic, Toxicology and Targeting, Research Institute of Pharmacy, University of Groningen, A. Deusinglaan 1, 9713AV Groningen, The Netherlands
eSchool of Chemistry, Cardiff University, Park Place, CF103AT Cardiff, UK. E-mail: casinia@cardiff.ac.uk; Fax: +44 (0)29 2087 4030; Tel: +44 (0)29 2087 6364
First published on 2nd February 2016
Abstract
The reaction of CoCl2·6H2O with the quinolone antimicrobial agent flumequine (Hflmq) in the absence or presence of the α-diimines 2,2′-bipyridine (bipy), 1,10-phenanthroline (phen) or 2,2′-bipyridylamine (bipyam) resulted in the formation of four mononuclear complexes which were characterized with physicochemical and spectroscopic techniques. The crystal structures of [Co(flmq)2(bipy)]·2H2O, [Co(flmq)2(phen)]·1.6MeOH·0.4H2O and [Co(flmq)2(bipyam)]·H2O were determined by X-ray crystallography. The interaction of the complexes with calf-thymus DNA (CT DNA) was investigated by UV spectroscopy, viscosity measurements, cyclic voltammetry and competitive studies with ethidium bromide in order to evaluate the possible DNA-binding mode and to calculate the corresponding DNA-binding constants. The binding of the complexes to human or bovine serum albumin was studied by fluorescence emission spectroscopy and the corresponding binding constants were determined. The antimicrobial activity of the Co(II)–flumequine and the recently reported Cu(II)–flumequine complexes was tested against four different microorganisms (Escherichia coli, Xanthomonas campestris, Staphylococcus aureus and Bacillus subtilis) and was found to be similar to that of free Hflmq. The antiproliferative activity of previously reported complexes [Cu(flmq)(phen)Cl], [Zn(flmq)(phen)Cl] and [Ni(flmq)2(phen)] against human ovarian (A2780) and lung (A549) cancer cell lines is also reported in comparison to the cobalt analogue, [Co(flmq)(phen)Cl], 3, highlighting important differences among the various complexes which may be due to different uptake and modes of action.
Introduction
Quinolones (quinolonecarboxylic acids or 4-quinolones) are synthetic antibacterial drugs bearing a 4-oxo-1,4-dihydroquinoline skeleton and are widely used for the treatment of many infections including urinary tract, respiratory and bone-joint infections, sexually transmitted diseases, prostatitis, pneumonia and acute bronchitis.1–3 For the time being, quinolones are among the most clinically successful antibacterial agents4 with the enzymes gyrases (type II topoisomerases) and topoisomerase IV that participate in DNA replication being their main biological targets.5–7 Microbial resistance to drugs is a big public health threat to the world nowadays. Various studies have been performed to explain and to solve this difficult issue. For example, quinazolinediones (diones), fluoroquinolone-like topoisomerase poisons that are unaffected by common quinolone-resistance mutations, were tested to bypass fluoroquinolone resistance.8 It is known that the most common causes of quinolone resistance are mutations of specific amino-acid residues in the gyrase or topoisomerase IV enzyme and these amino-acids are proposed to serve as a critical enzyme–quinolone interaction site. Metal ions, especially magnesium (but also others were tested), are involved in these processes. Serine and glutamic acid residues act by anchoring a water–metal ion bridge that coordinates drug binding.9 Results of various studies also suggest that the cell intake route of free quinolone is different from that of quinolone–metal complexes.10 This supports the suitability of metal complexes as candidates for further biological testing in quinolone resistant microorganisms. Within this context, the interaction of quinolones as well as their metal complexes with various types of DNA has been studied as a means to better explain their antibacterial activity on diverse microorganisms.11–13 Furthermore, these compounds have been tested for antibacterial activity on diverse microorganisms,14–18 and their cytotoxicity and potential antitumor activity19–27 with metal complexes of the drugs being, in many cases, more active than their parent compounds.2,28Flumequine (Hflmq) is a first-generation quinolone and is structurally related to ofloxacin, nalidixic acid and oxolinic acid.29,30 Hflmq is chiral and a racemic mixture (Fig. 1) is used as a ligand. It is highly potent for the treatment of urinary tract infections, since it is active against some Gram-positive and Gram-negative microorganisms.31,32 Diverse nickel(II),33 copper(II)34 and zinc(II)35,36 complexes with flumequine as ligand have been structurally characterized and recently reported.28
 | ||
| Fig. 1 The racemic mixture of flumequine (=Hflmq). | ||
The most well-known and most important biological role of cobalt is its presence in the active center of vitamin B12; as a result, cobalt is considered to participate indirectly in the regulation of DNA synthesis.37 Cobalt is also used as a supplement to the vitamin B12 and is related to more than eight cobalt-dependent proteins.37,38 In the last six decades, the interest of researchers in regard to the biological activity of cobalt compounds has been expanding.39 More specifically, the cobalt complex doxovir (or CTC-96) has shown antipsoriatic activity and has already completed successfully phase II clinical trials for the treatment of herpes simplex virus.40 Furthermore, diverse cobalt compounds have shown antibacterial,41–43 antifungal,44,45 antioxidant,46,47 antiproliferative48,49 and antiviral50,51 activity and others have been reported for hydrolytic cleavage and binding of DNA.52 A thorough research of the literature in regard to cobalt–quinolone complexes has revealed that Co(II) complexes with the quinolones ciprofloxacin,17 enoxacin,53 enrofloxacin,18 oxolinic acid,43 norfloxacin and sarafloxacin54 have been structurally characterized.
As a continuation of our recent research involving the interaction of cobalt(II) with quinolones,18,43 we report herein the synthesis, characterization and biological properties of cobalt(II) complexes with the first-generation quinolone flumequine and the oxygen-donor methanol or the nitrogen-donors 2,2′-bipyridine (bipy), 1,10-phenanthroline (phen) and 2,2′-bipyridylamine (bipyam) as co-ligands. The synthesized compounds bear the formulas [Co(flmq)2(MeOH)2], 1 and [Co(flmq)2(L)], (L = bipy, phen or bipyam) for complexes 2–4, respectively. The complexes were characterized by physicochemical and spectroscopic techniques and the crystal structures of [Co(flmq)2(bipy)]·2H2O, 2·2H2O, [Co(flmq)2(phen)]·1.6MeOH·0.4H2O, 3·1.6MeOH·0.4H2O and [Co(flmq)2(bipyam)]·H2O, 4·H2O were determined by X-ray crystallography. The interaction of complexes 1–4 with calf-thymus (CT) DNA has been investigated by UV spectroscopy, cyclic voltammetry, viscosity measurements and by determining their ability to displace the well-known DNA-intercalator ethidium bromide (EB) from its CT DNA–EB complex by fluorescence emission spectroscopy, in order to conclude the mode and strength of binding. The affinity of complexes 1–4 to bind to bovine (BSA) and human (HSA) serum albumins, proteins involved in their potential transportation through the bloodstream, was investigated by fluorescence emission spectroscopy. The antimicrobial activity of the cobalt(II)–flumequine complexes 1–4 and the previously reported copper(II)–flumequine complexes [Cu(flmq)2(H2O)2], 5, [Cu(flmq)(bipy)Cl], 6, [Cu(flmq)(phen)Cl], 7, [Cu(flmq)(bipyam)Cl], 8 and [Cu(flmq)2(py)2], 9 (py = pyridine)34 was evaluated by determining the half-minimum inhibitory concentration (IC50) and the minimum inhibitory concentration (MIC) against four Gram-positive or Gram-negative microorganisms (i.e. Escherichia coli NCTC 29212 (E. coli) Xanthomonas campestris ATCC 1395 (X. campestris), Staphylococcus aureus ATCC 6538 (S. aureus) and Bacillus subtilis ATCC 6633 (B. subtilis)). Additionally, the antiproliferative activity of recently reported complexes [Cu(flmq)(phen)Cl], 7,34 [Zn(flmq)(phen)Cl], 10 (ref. 36) and [Ni(flmq)2(phen)], 11 (ref. 33) was examined against human ovarian (A2780) and lung (A549) cancer cell lines.
Experimental
Materials – instrumentation – physical measurements
All chemicals (CoCl2·6H2O, flumequine, phen, bipy, bipyam, KOH, CT DNA, BSA, HSA, EB, NaCl, trisodium citrate) were purchased from Sigma-Aldrich and all solvents were purchased from Chemlab. All the chemicals and solvents were reagent grade and were used as purchased without any further purification.DNA stock solution was prepared by dilution of CT DNA with buffer (containing 15 mM trisodium citrate and 150 mM NaCl at pH 7.0) followed by exhaustive stirring for three days, and was kept at 4 °C for no longer than a week. The stock solution of CT DNA gave a ratio of UV absorbance at 260 and 280 nm (A260/A280) of ∼1.86, indicating that DNA was sufficiently free of protein contamination.55 The DNA concentration was determined by the UV absorbance at 260 nm after 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 dilution using ε = 6600 M−1 cm−1.56
:20 dilution using ε = 6600 M−1 cm−1.56
Infrared (IR) spectra (400–4000 cm−1) were recorded on a Nicolet FT-IR 6700 spectrometer with samples prepared as KBr disks. UV-visible (UV-vis) spectra were recorded as nujol mulls and in DMSO solution at concentrations in the range 10−5 to 5 × 10−3 M on a Hitachi U-2001 dual beam spectrophotometer. Room temperature magnetic measurements were carried out by the Faraday method. C, H and N elemental analyses were performed on a Perkin-Elmer 240B elemental analyzer. Molar conductivity measurements of 1 mM DMSO solution of the complexes were carried out with a Crison Basic 30 conductometer. Viscosity experiments were carried out using an ALPHA L Fungilab rotational viscometer equipped with an 18 mL LCP spindle and the measurements were performed at 100 rpm. Fluorescence spectra were recorded in solution on a Hitachi F-7000 fluorescence spectrophotometer.
Cyclic voltammetry studies were performed on an Eco Chemie Autolab Electrochemical analyzer. Cyclic voltammetry experiments were carried out in a 30 mL three-electrode electrolytic cell. The working electrode was a platinum disk, a separate Pt single-sheet electrode was used as the counter electrode and a Ag/AgCl electrode saturated with KCl was used as the reference electrode. Oxygen was removed by purging the solutions with pure nitrogen which had been previously saturated with solvent vapours. All electrochemical measurements were performed at 25.0 ± 0.2 °C.
Synthesis of the complexes
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)pyridone, 1621 (vs (very strong)); νasym(CO2), 1585 (vs); νsym(CO2), 1377 (s (strong)); Δν(CO2) = νasym(CO2) − νsym(CO2): 208 cm−1; UV-vis: as nujol mull, λ/nm: 642 (sh (shoulder)), 528, 422 (sh), 392, 339 (sh), 326; in DMSO, λ/nm (ε/M−1 cm−1): 640 (sh) (10), 526 (40), 420 (70), 386 (550), 341 (4850), 329 (5100), 309 (5600). μeff at room temperature = 3.94 BM. The complex is soluble in DMSO (ΛM = 5 S cm2 mol−1, in 1 mM DMSO solution) and partially soluble in methanol, acetonitrile and DMF.
O)pyridone, 1621 (vs (very strong)); νasym(CO2), 1585 (vs); νsym(CO2), 1377 (s (strong)); Δν(CO2) = νasym(CO2) − νsym(CO2): 208 cm−1; UV-vis: as nujol mull, λ/nm: 642 (sh (shoulder)), 528, 422 (sh), 392, 339 (sh), 326; in DMSO, λ/nm (ε/M−1 cm−1): 640 (sh) (10), 526 (40), 420 (70), 386 (550), 341 (4850), 329 (5100), 309 (5600). μeff at room temperature = 3.94 BM. The complex is soluble in DMSO (ΛM = 5 S cm2 mol−1, in 1 mM DMSO solution) and partially soluble in methanol, acetonitrile and DMF.
[Co(flmq)2(bipy)]·2H2O, 2·2H2O. Orange crystals of [Co(flmq)2(bipy)]·2H2O, 2·2H2O (100 mg, yield: 65%) suitable for X-ray structure determination, were collected after twenty days. Anal. calcd for [Co(flmq)2(bipy)]·2H2O (C38H34CoF2N4O8) (MW = 771.62): C 59.15%, H 4.44, N 7.26; found C 59.28, H 4.28, N 7.06; IR (KBr pellet): νmax/cm−1ν(C
O)pyridone: 1636 (vs); νasym(CO2): 1587 (vs); νsym(CO2): 1373 (s); Δν(CO2) = 214 cm−1; UV-vis: as nujol mull, λ/nm: 638, 532, 428, 394, 342 (sh), 329; in DMSO, λ/nm (ε/M−1 cm−1): 640 (sh) (5), 530 (35), 425 (120), 395 (600), 341 (8100), 329 (8600), 307 (7500). μeff at room temperature = 4.17 BM. The complex is soluble in DMSO (ΛM = 8 S cm2 mol−1, in 1 mM DMSO solution) and partially soluble in DMF.
[Co(flmq)2(phen)]·1.6MeOH·0.4H2O, 3·1.6MeOH·0.4H2O. Red–orange crystals of [Co(flmq)2(phen)]·1.6MeOH·0.4H2O, 3·1.6MeOH·0.4H2O (100 mg, yield: 60%) suitable for X-ray structure determination, were collected after ten days. Anal. calcd for [Co(flmq)2(phen)]·1.6MeOH·0.4H2O (C41.6H37.2CoF2N4O8) (MW = 818.08): C 61.08, H 4.58, N 6.85; found C 61.44, H 4.41, N 6.51; IR (KBr pellet): νmax/cm−1ν(C
O)pyridone 1622 (vs); νasym(CO2): 1589 (vs); νsym(CO2): 1373 (v); Δν(CO2) = 216 cm−1; UV-vis: as nujol mull, λ/nm: 650 (sh), 525, 435 (sh), 392, 328; in DMSO, λ/nm (ε/M−1 cm−1): 645 (sh) (5), 520 (20), 430 (120), 390 (700), 339 (7600), 327 (8700), 305 (9600). μeff at room temperature = 4.22 BM. The complex is soluble in DMF and DMSO (ΛM = 9 S cm2 mol−1, in 1 mM DMSO solution).
[Co(flmq)2(bipyam)]·H2O, 4·H2O. Orange crystals of [Co(flmq)2(bipyam)]·2H2O, 4·2H2O (105 mg, yield: 70%) suitable for X-ray structure determination, were collected after two weeks. Anal. calcd for [Co(flmq)2(bipyam)]·H2O (C38H33CoF2N5O7) (MW = 768.62): C 59.38, H 4.33, N 9.11; found C 59.10, H 4.52, N 8.97; IR (KBr pellet): νmax/cm−1ν(C
O)pyridone 1622 (vs); νasym(CO2): 1583 (vs); νsym(CO2): 1373 (vs); Δν(CO2) = 210 cm−1; UV-vis: as nujol mull, λ/nm: 650 (sh), 525 (sh), 435, 390, 333; in DMSO, λ/nm (ε/M−1 cm−1): 645 (sh) (5), 520 (65), 430 (160), 391 (550), 339 (4100), 323 (6500), 305 (7000). μeff at room temperature = 4.35 BM. The complex is soluble in DMF and DMSO (ΛM = 10 S cm2 mol−1, in 1 mM DMSO solution).
X-ray structure determination
Single-crystal X-ray diffraction data were collected at room temperature on a Nonius Kappa CCD and Agilent Technologies SuperNova Dual diffractometers using Mo-Kα radiation (λ = 0.71073 Å). The data were processed using DENZO57 or CrysAlis Pro.58 The structures were solved by direct methods implemented in SIR97 (ref. 59) or SHELXLS-97 (ref. 60) and refined by a full-matrix least-squares procedure based on F2 with SHELXL-97.60 All the non-hydrogen atoms were refined anisotropically. Hydrogen atoms were readily located in difference Fourier maps and were subsequently treated as riding atoms in geometrically idealized positions with Uiso(H) = kUeq(C, N or O), where k = 1.5 for OH and methyl groups, which were permitted to rotate but not to tilt, and 1.2 for all other H atoms unless otherwise noted.In the crystal structure of 2, the flumequine ligands have a disorder over two positions at C12 and C14 and at C26 and C28 with a refined occupancy in ratios 0.61:0.39 and 0.55:0.45, respectively. Hydrogen atoms attached to water oxygen atoms O7 and O8 were refined fixing the bond lengths. In the crystal structure of 3, the flumequine ligand has a disorder over two positions at C12 and C14 with a refined occupancy in ratios 0.50:0.50. The C10 and C11 atoms were refined using SIMU and DELU instructions, C11 also using ISOR instruction. Crystals were isolated as mixed methanol/water solvate, with MeOH:H2O refined ratio 0.80:0.20. Hydrogen atoms on water oxygen atom O5 were not found in difference Fourier maps and were not included in the refinement. In the crystal structure of 4, the water oxygen atom O7 has a disorder over two positions in ratio 0.72:0.28. Hydrogen atoms on water oxygen atom O7 were not found in difference Fourier maps and were not included in the refinement. Crystallographic data are listed in Table 1.
| 2·2H2O | 3·1.6MeOH·0.4H2O | 4·H2O | |
|---|---|---|---|
| Empirical formula | C38H34CoF2N4O8 | C41.6H37.2CoF2N4O8 | C38H33CoF2N5O7 |
| M w | 771.62 | 818.08 | 768.62 |
| T, K | 293(2) | 293(2) | 293(2) |
| Crystal system | Triclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P2/c | P21/n |
| a (Å) | 9.2333(2) | 11.4479(4) | 11.5948(10) |
| b (Å) | 9.5588(3) | 10.0270(3) | 24.2372(15) |
| c (Å) | 20.9349(5) | 18.5359(7) | 12.5448(9) |
| α (°) | 98.156(2) | 90.00 | 90.00 |
| β (°) | 102.649(2) | 114.920(3) | 101.391(8) |
| γ (°) | 96.981(2) | 90.00 | 90.00 |
| V (Å3) | 1762.20(8) | 1929.60(11) | 3456.0(4) |
| Z | 2 | 2 | 4 |
| D calc (g cm−3) | 1.454 | 1.408 | 1.477 |
| μ (mm−1) | 0.557 | 0.513 | 0.567 |
| Data collected/unique/[I > 2σ(I)] | 14676/7981/6268 |
18417/4412/3356 |
18382/7890/5212 |
| Restraints/parameters | 4/536 | 13/291 | 0/491 |
| S | 1.005 | 1.086 | 1.035 |
| R 1, wR2 [I > 2σ(I)] | 0.0414/0.1064 | 0.0612/0.1766 | 0.0655/0.1676 |
| R 1, wR2 (all data) | 0.0590/0.1169 | 0.0754/0.1887 | 0.1013/0.1914 |
| Largest diff. peak/hole (e Å−3) | 0.268/−0.344 | 0.749/−0.282 | 0.993/−0.440 |
DNA-binding studies
:2 DMSO:buffer solution of complex upon addition of DNA. The buffer was used as the supporting electrolyte and the cyclic voltammograms were recorded at ν = 100 mV s−1.
Albumin binding studies
The albumin binding study was performed by tryptophan fluorescence quenching experiments using bovine (BSA, 3 μM) or human serum albumin (HSA, 3 μM) in buffer (containing 15 mM trisodium citrate and 150 mM NaCl at pH 7.0). The quenching of the emission intensity of tryptophan residues of BSA at 343 nm or HSA at 351 nm was monitored using complexes 1–4 as quencher with increasing concentration. The fluorescence emission spectra were recorded from 300 to 500 nm at an excitation wavelength of 295 nm.63 The fluorescence emission spectra of complexes 1–4 in buffer solutions were recorded under the same experimental conditions (i.e. excitation at 295 nm) and exhibited a maximum emission at 365 nm.33,34 Therefore, quantitative studies of the serum albumin fluorescence emission spectra were performed after their correction by subtracting the spectra of the compounds. The influence of the inner-filter effect on the measurements was evaluated by eqn (S3).†64 The Stern–Volmer and Scatchard equations (eqn (S4)–(S6)†)65 and graphs have been used in order to study the interaction of each quencher with the serum albumins and calculate the dynamic quenching constant KSV (in M−1), the approximate quenching constant kq (in M−1 s−1), the SA-binding constant K (in M−1) and the number of binding sites per albumin n.Determination of the antimicrobial activity
The antimicrobial activity of Hflmq and its cobalt(II) and copper(II) complexes was evaluated by determining their respective IC50 and MIC values towards two Gram-(−) (E. coli and X. campestris) and two Gram-(+) (S. aureus and B. subtilis) bacterial species. Cultures of these microbial strains were grown on a rich selective agar medium and stored at 4 °C. The selective media used were nutrient agar or broth for B. subtilis and S. aureus, yeast mold agar or broth for X. campestris and Luria agar or broth for E. coli. Cells picked from the surface of the stored cultures were used to initiate liquid pre-cultures of the same selective medium at an initial turbidity of roughly 1 McFarland unit. Pre-cultures were incubated for 24 h in a rotary shaking incubator and subsequently they were used to inoculate the test cultures used for the determination of MIC at an initial turbidity of 0.5 McFarland units. The test cultures consisted of Mueller-Hinton broth (Deben Diagnostics Ltd) containing different concentrations of the compounds. Different concentrations were achieved as follows: the compounds were freshly dissolved in DMSO to a concentration of 1 mg mL−1 and they were diluted with DMSO, using the method of progressive double dilution. Thus, working solutions with decreasing concentrations of the compounds under investigation were achieved. The working solutions were subsequently diluted to the final desired concentration by addition to the growth medium at a proportion of 2:98. MIC values were determined as the lowest concentrations of the tested compounds that inhibited visible growth of each respective organism after a 24 h incubation.66 Bacterial growth was determined by measuring the turbidity of appropriately diluted cultures at 600 nm with reference to equally diluted sterile growth medium and the inhibition achieved was calculated by comparing the turbidity of each culture to the average of the turbidity of three non-inhibited cultures. The IC50 values were calculated using a linear regression equation of the inhibitory effects of at least three concentrations equal or higher than the MIC and the decimal logarithm of the concentration.67 All test cultures were grown in triplicate and for the determination of MIC, growth had to be inhibited in at least two cultures of the triplicate. The incubation temperature at all stages was 37 °C except for X. campestris which was cultivated at 28 °C.
The effect on the growth of E. coli was monitored for 48 h using a modification of previously described methods68,69 in sterile 96-well flat bottom microtitre plates closed with sterile standard-profile lids without condensation rings. The inoculum of the test cultures was prepared in the same manner as for the determination of the MIC with the difference that the initial turbidity of 0.5 McFarland units was achieved by mixing 20 μL of the antibiotic solution with 230 μL of the appropriately inoculated Mueller-Hinton broth in each microtitre well. The covered microtitre plates were placed in a Synergy 2 Multi-Mode Reader (BioTek, USA), incubated at 35 °C for 48 h with no shaking and the absorbance at 600 nm was measured every 20 min.
Antiproliferative assay in cancer cells
The human lung cancer (A549) and human ovarian cancer sensitive to cisplatin (A2780) cell lines (obtained from the European Centre of Cell Cultures ECACC, Salisbury, UK) were cultured in DMEM (for A549) or RPMI (for A2780) both containing GlutaMax-I supplemented with 10% FBS and 1% penicillin/streptomycin (all from Invitrogen), at 37 °C in a humidified atmosphere of 95% of air and 5% CO2 (Heraeus, Germany). For evaluation of growth inhibition, cells were seeded in 96-well plates (Costar, Integra Biosciences, Cambridge, MA) at a concentration of 104 cells per well and grown for 24 h in complete medium. Solutions of the compounds were prepared by diluting a freshly prepared stock solution (10−2 M in DMSO) of the corresponding compound in aqueous media (RPMI or DMEM depending on the cell lines). The percentage of DMSO in the culture medium never exceeded 0.2%: at this concentration DMSO has no effect on the cell viability. Cisplatin (Sigma-Aldrich) stock solutions were freshly prepared in aqueous solutions. Afterwards, the intermediate dilutions of the compounds were added to the wells (200 μL) to obtain a final concentration ranging from 0 to 200 μM, and the cells were incubated for 72 h. Following 72 h drug exposure, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to the cells at a final concentration of 0.50 mg mL−1 incubated for 3–4 h, then the culture medium was removed and the violet formazan dissolved in DMSO. The optical density of each well (96-well plates) was quantified in quadruplicate at 540 nm using a multi-well plate reader and the percentage of surviving cells was calculated from the ratio of absorbance between treated and untreated cells. The IC50 value was calculated as the concentration reducing the proliferation of the cells by 50% and is presented as a mean (±SE) of at least three independent experiments.Cell uptake studies and ICP-MS analysis
For the evaluation of the cell uptake, cells were seeded in 6-well plates and grown to approximately 70% confluency and incubated with compound 3 at 100 μM for 24 h. At the end of the incubation period, cells were rinsed with 5 mL of PBS, detached by adding 0.4 mL enzyme free cell dissociation solution (Millipore) and collected by centrifugation. Cellular extracts were prepared according to established procedures.70All samples were analyzed for their protein content (to establish the number of cells per sample) prior to ICP-MS determination using a BCA assay (Sigma Aldrich). All samples were digested in ICP-MS grade concentrated hydrochloric acid (Sigma Aldrich) for 3 h at room temperature and filled to a total volume of 8 mL with ultrapure water. Indium was added as an internal standard at a concentration of 0.5 ppb. Determinations of total metal contents were achieved on an Elan DRC II ICP-MS instrument (Perkin Elmer). The ICP-MS instrument was tuned daily using a solution provided by the manufacturer containing 1 ppb each of Mg, In, Ce, Ba, Pb and U. External standards were prepared gravimetrically in an identical matrix to the samples (with regard to internal standard and hydrochloric acid) with single element standards obtained from CPI International (Amsterdam, The Netherlands). The results are expressed as mean ± SE of at least three determinations.
Discussion
Synthesis and characterization of complexes 1–4
The complexes were synthesized in high yield via the aerobic reaction of flumequine, deprotonated by KOH, with CoCl2·6H2O in the absence (for 1) or presence of the corresponding N,N′-donor co-ligand (L = bipy, phen, bipyam) for 2–4, according to the following equations:| CoCl2·6H2O + 2Hflmq + 2KOH + 2MeOH → [Co(flmq)2(MeOH)2] + 2KCl + 8H2O | (1) |
| CoCl2·6H2O + 2Hflmq + 2KOH + L → [Co(flmq)2(L)] + 2KCl + 8H2O | (2) |
The complexes were characterized by elemental analysis, IR and UV-vis spectroscopic techniques, magnetic measurements at room temperature and, in the case of 2–4, by X-ray crystallography.
The composition of complex 1 is Co:flmq:MeOH = 1:2:2, while complexes 2–4 have a 1:2:1 Co:flmq:L composition (L = bipy, phen, bipyam), as is indicated from elemental analysis. Complexes 1–4 are soluble in DMSO and are not electrolytes; the values of the molar conductivity of 1 mM DMSO solution of the complexes (ΛM = 5–10 S cm2 mol−1) might suggest a slight partial ionization, but the dissociation degree is very low (for a 1:1 electrolyte, the ΛM value should be ∼70 S cm2 mol−1) and, thus, we may consider that the compounds do not dissociate in DMSO solution.43
The IR spectra of the complexes were recorded in order to confirm the deprotonation and the binding mode of flumequine. In the IR spectrum of free Hflmq, the bands located at 3435 (broad, medium) cm−1, 1718 (s (strong)) cm−1 and 1270 (s) cm−1 were attributed the ν(O–H), ν(CO)carboxyl and ν(C–O)carboxyl stretching vibrations of the carboxyl group (–COOH) and at 1618 (vs) cm−1 attributed to ν(CO)pyridone stretching vibration.33,34 In the IR spectra of the cobalt(II) complexes 1–4, the ν(O–H) disappeared indicating the deprotonation of the carboxylate group upon binding to metal ion, and the ν(CO)carboxyl and ν(C–O)carboxyl shifted towards the range 1583–1589 (vs) cm−1 and 1373–1377 (s) cm−1 and were characterized as antisymmetric, νasym(CO2), and symmetric, νsym(CO2), stretching vibrations of the carboxylato group, respectively. The values of parameter Δν(CO2) [=νasym(CO2) − νsym(CO2)] are in the range 208–216 cm−1 and suggest the monodentate coordination mode of the carboxylate group of the quinolone ligand.71 Furthermore, the ν(CO)pyridone shifted up to 1621–1636 cm−1 as a result of its coordination. All these spectral features are characteristic of coordination of the deprotonated flumequine ligands to cobalt in a chelating bidentate mode via the pyridone oxygen and a carboxylato oxygen.33–36
The UV-vis spectra of the complexes were recorded as nujol mulls and in DMSO solution and present similar patterns, suggesting thus that the complexes retain their structure in solution. In particular, three low-intensity bands due to d–d transitions were observed in the visible spectra of the complexes being characteristic for distorted octahedral high-spin Co2+ complexes;37 namely, band I in the range 640–650 nm (ε = 5–10 M−1 cm−1) assigned to 4T1g(F) → 4T2g transition, band II at 520–530 nm (ε = 20–65 M−1 cm−1) assigned to 4T2g(F) → 4A2g transition and band III at 420–430 nm (ε = 70–160 M−1 cm−1) to 4T1g(F) → 4T1g(P) transition. Furthermore, the band located in the range 386–395 nm (ε = 550–700 M−1 cm−1) was assigned to charge-transfer transition as observed in previously reported metal–quinolone complexes.18,43
The UV-vis spectra of 1–4 were also recorded in the presence of a series of buffer solutions in the pH range 6–8 (150 mM NaCl and 15 mM trisodium citrate at pH values regulated by HCl solution) in order to examine whether the complexes are stable in the buffer solution used for the biological experiments. Since no significant changes (i.e. shift of the λmax or new peaks) were observed, the integrity of the complexes in the presence of the buffer solution used for the biological experiments may be suggested. When the observations derived for UV-vis spectroscopy (similarity of the spectra in nujol, in DMSO solution and in mixtures of DMSO/buffer solution) are combined with the non-electrolytic nature of the complexes as shown by molar conductivity measurements, we may conclude that complexes 1–4 keep their integrity in solution.18,43
The observed values of the magnetic moment at room temperature for complexes 1–4 (μeff = 3.94–4.35 BM) are higher than the spin-only value (=3.87 BM), show a spin–orbit coupling which is probably due to the t52ge2g electron configuration and are characteristic of mononuclear high-spin Co(II) complexes (S = 3/2).18,37,43
Structures of the complexes
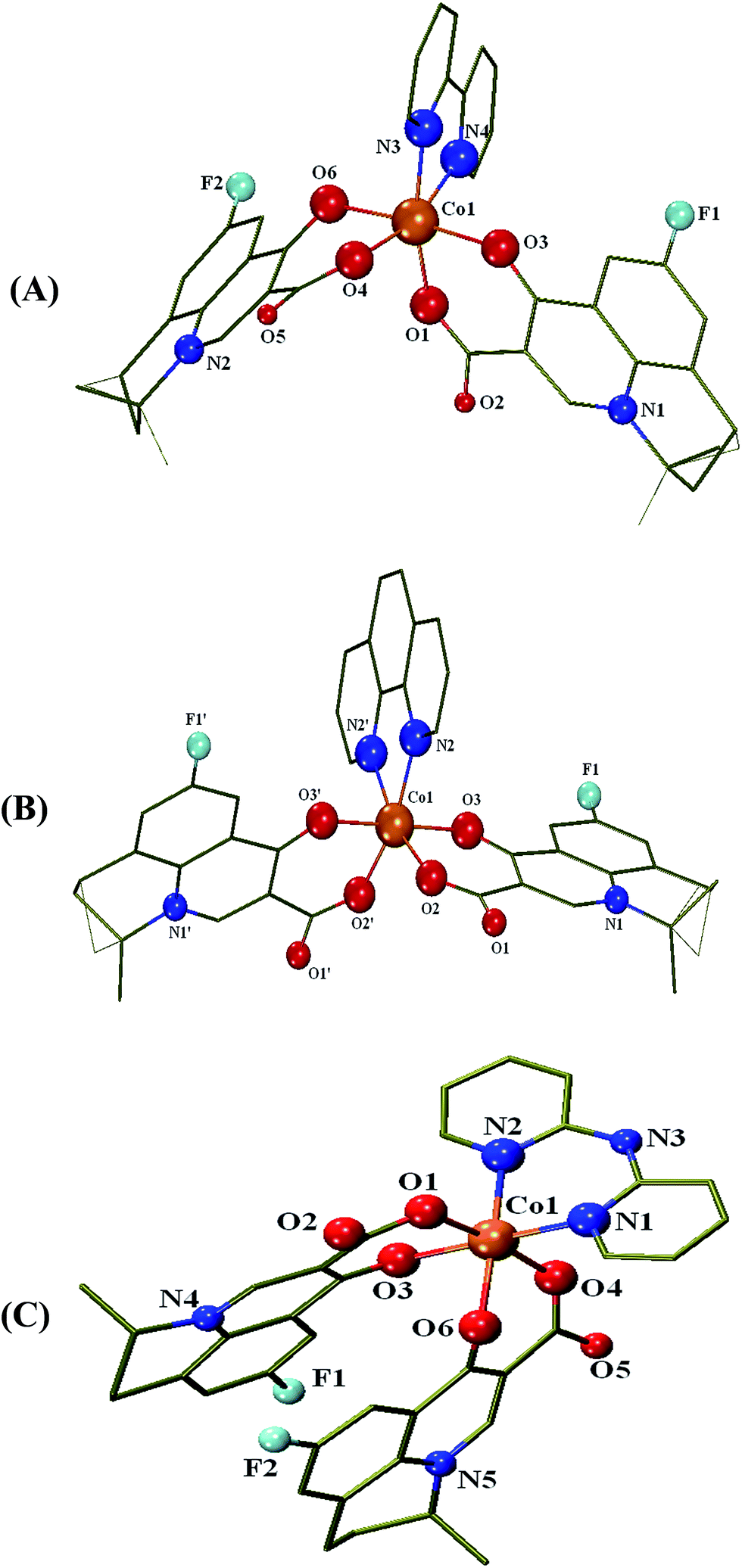 | ||
| Fig. 2 Partially labelled plot of the molecular structure of complex (A) [Co(flmq)2(bipy)], 2 (B) [Co(flmq)2(phen)], 3 and (C) [Co(flmq)2(bipyam)], 4. Hydrogen atoms and disorder atoms are omitted for clarity. | ||
| Bond | [Co(flmq)2(bipy)] | [Co(flmq)2(phen)] | [Co(flmq)2(bipyam)] |
|---|---|---|---|
| Distance (Å) | Distance (Å) | Distance (Å) | |
| Co–Ocarb | 2.0302(14), 2.0326(16) | 2.028(2) | 2.023(3), 2.052(2) |
| Co–Opyr | 2.0852(14), 2.0890(13) | 2.073(2) | 2.097(2), 2.136(2) |
| Co–N | 2.1256(15), 2.1300(18) | 2.152(2) | 2.110(3), 2.112(3) |
| C–Ocarb,bound | 1.253(2), 1.265(3) | 1.254(3) | 1.260(5), 1.266(4) |
| C–Ocarb,unbound | 1.232(2), 1.247(3) | 1.244(4) | 1.236(5), 1.237(4) |
| C–Opyr | 1.259(2), 1.260(2) | 1.260(4) | 1.266(4), 1.261(4) |
| Bond angle | Angle (°) | Angle (°) | Angle (°) |
|---|---|---|---|
| Ocarb,1–Co–Ocarb,2 | 98.45(7) | 99.71(14) | 173.55(10) |
| Ocarb,1–Co–Opyr,1 | 86.52(6), 86.93(5) | 86.63(9) | 86.61(10), 85.56(10) |
| Ocarb,1–Co–Opyr,2 | 92.15(7), 88.15(6) | 92.79(9) | 88.47(11), 90.27(10) |
| Ocarb–Co–N | 93.52(7), 166.17(7), 93.55(6), 163.85(7) | 92.11(9), 166.43(9) | 89.11(12), 92.14(11), 93.67(12), 94.02(12) |
| Opyr–Co–Opyr | 174.66(5) | 179.11(13) | 83.27(10) |
| Opyr–Co–N | 83.04(6), 85.78(6), 99.33(7), 99.46(6) | 94.55(9), 86.15(9) | 93.27(11), 96.79(11), 175.83(11), 179.37(12) |
| N–Co–N | 76.13(6) | 77.04(13) | 86.63(12) |
The Co(II) atom is six-coordinated exhibiting a distorted octahedral geometry and four oxygen atoms from two flumequine ligands and two nitrogen atoms from the bidentate N,N′-donor co-ligand (bipy, phen, bipyam) occupy the six vertices of the octahedron. The bond distances around the Co(II) are quite different; the Co–Ocarb distances [=2.023(3)–2.052(2) Å] are slightly shorter than Co–Opyr [=2.073(2)–2.136(2) Å] while the Co–N [=2.110(3)–2.152(2) Å] distances are the longest ones.
In the structures of complexes 2 and 3, the two carboxylato oxygen atoms are cis to each other [Ocarb–Co–Ocarb = 98.45(7)° and 99.71(14)°, respectively] and the two pyridone oxygen atoms [Opyr–Co–Opyr = 174.66(5)° and 179.11(13)°, respectively] are in a trans arrangement. On the other hand, in the crystal structure of complex 4, the arrangement of the flumequine oxygen atoms around Co(II) is different with the pyridone oxygens [Opyr–Co–Opyr = 83.27(10)°] being in a cis arrangement and the carboxylato oxygen [Ocarb–Co–Ocarb = 173.55(10)°] lying trans to each other. In the reported quinolone complexes of the formula [M(Q)2(N,N′-donor)], all three different arrangement modes of quinolone coordinated oxygens around the metal ion have been observed:28 (i) the carboxylato oxygens lying at cis positions and pyridone oxygens at trans positions as in complexes 2 and 3 as well as in [Zn(flmq)2(bipy)],35 [Zn(flmq)2(phen)],36 [Ni(flmq)2(phen)]33 and [Co(erx)2(bipyam)],18 (ii) the pyridone oxygens at cis positions and the carboxylato oxygens at trans positions as in complex 4 and [Ni(flmq)2(bipy)]33 and (iii) the carboxylato and the pyridone oxygens at cis positions as in a series of Ni(II)–quinolone complexes.28
The N,N′-donor ligand is almost planar with the cobalt atom and the N–Co–N angles [=76.13(6)–86.63(12)°] are within the range of reported values for complexes containing chelating polycyclic α-diimines.46,72,73
In the crystal structure of 2·2H2O, the solvate water molecules enable the formation of a 1D hydrogen-bonded chain (Table S4 and Fig. S1†). The structure is further stabilized by π⋯π [3.7699(19) Å, 4.0082(13) Å and 4.1354(16) Å] (Fig. S1†) and weak CH⋯π, CF⋯π and CH⋯O interactions. In the crystal structure of 3·1.6MeOH·0.4H2O, a solvate methanol molecule is hydrogen-bonded to the complex 3 (Table S4†) and the structure is stabilized by π⋯π interactions [3.688(3) Å] (Fig. S2†) as well as by weak CH⋯π and CF⋯π interactions. While in 2·2H2O and 3·1.6MeOH·0.4H2O the bipy and phen ligands, respectively, are not involved in any significant π⋯π interactions and flmq ligands only to some extent, in 4·H2O all aromatic rings participate in this kind of interaction. The intramolecular π⋯π interactions have a role in the stabilization of the (ii) arrangement mode of molecular structure of 4 [3.818(2) Å, 3.895(2) Å, 4.147(2) Å and 4.326(2) Å]. Additionally, a stack is formed through the intermolecular π⋯π interactions between flmq ligands of adjacent molecules [3.824(2) Å]. π⋯π interactions between bipyam ligands of adjacent molecules [4.123(3) Å] are enhancing the hydrogen-bonded chain formed via N–H⋯O hydrogen bonds involving the bipyam NH-group and the carboxylic oxygen atom of the flmq ligand due to the presence of water molecules (Table S4 and Fig. S3†). The structure is further stabilized also by weak CF⋯π interactions.
Interaction of the complexes with CT DNA
Quinolones are compounds that can act as antibacterial agents because they are involved in the inhibition of DNA-replication since their targets are the bacterial topoisomerases DNA-gyrase (topoisomerase II) and topoisomerase IV.1–3 Therefore, the investigation of the interaction of quinolones and their complexes with DNA is of great interest as a first step of potential activity. In general, the binding of metal complexes to double-stranded DNA occurs via a covalent or noncovalent mode; in the noncovalent mode, the intercalation of the complex in-between the DNA nucleobases via π → π stacking, the groove-binding along the grooves of the DNA helix via van der Waals interaction or hydrogen-bonding or hydrophobic bonding and the electrostatic interactions due to Coulomb forces between metal complexes and the phosphate groups of DNA are included.74A technique that may provide useful preliminary information concerning the interaction mode and the binding strength of the compounds with DNA is the UV spectroscopic titration. Initially, the UV spectra of a CT DNA solution (C = 1.2 to 1.6 × 10−4 M) were recorded in the presence of complexes 1–4 at increasing amounts (for different [complex]/[DNA] mixing ratios (=r)). The slight decrease of the intensity at λmax = 258 nm observed in the UV spectra of a CT DNA solution (1.57 × 10−4 M) in the presence of increasing amounts of complex 2, as shown representatively in Fig. 3(A), indicates that the interaction of CT DNA with the complex results in the formation of a new complex with double-helical CT DNA.75,76 Quite similar behaviour of CT DNA is observed in the presence of the other complexes.
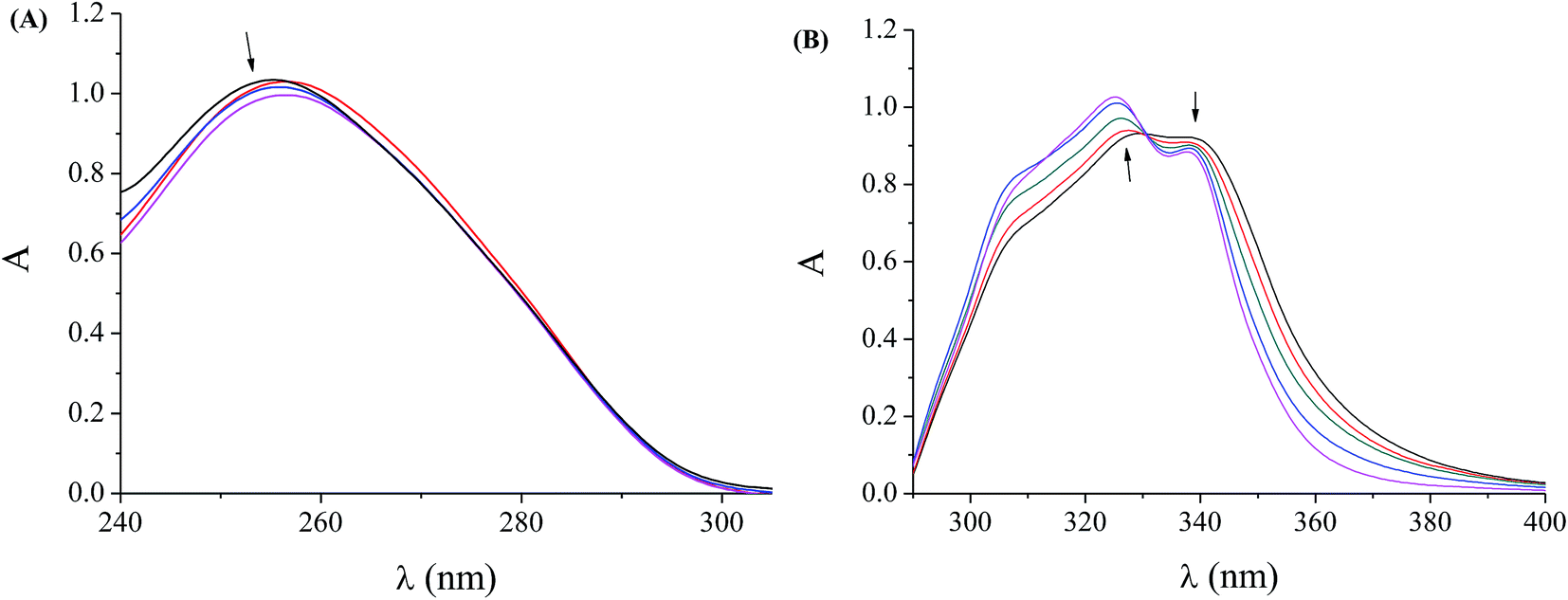 | ||
| Fig. 3 (A) UV spectra of CT DNA (1.57 × 10−4 M) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0) in the absence or presence of [Co(flmq)2(bipy)], 2. The arrow shows the changes upon increasing amounts of the complex (up to r = 0–0.6). (B) UV spectra of DMSO solution (5 × 10−5 M) of [Co(flmq)2(bipy)], 2 in the presence of increasing amounts of CT DNA. The arrows show the changes upon increasing amounts of CT DNA (r′ = 0–0.8). | ||
As a next step, the UV spectra of a DMSO solution of complexes 1–4 (2.5 to 5 × 10−5 M) were recorded in the presence of CT DNA at diverse r values; any interaction between the complex and CT DNA may perturb the intra-ligand transition bands of the complexes during the titrations.77 In the UV spectra of complex 2 (5 × 10−5 M), the intraligand bands at 329 nm (band I) and at 338 nm (band II) present a slight hyperchromism and slight hypochromism, respectively, upon addition of increasing amounts of CT DNA (up to r′ = [DNA]/[complex] = 0.8), (Fig. 3(B) for (r′ = 0–0.8)) followed by the appearance of an isosbestic point at 331 nm. Quite similar behaviour is observed for complexes 1, 3 and 4 upon the addition of CT (Table 3). In general, the observed hypochromism could be attributed to a π → π* stacking interaction between the aromatic chromophore (from flumequinato and/or the N-donor ligands) of the complex and DNA base pairs consistent with the intercalative binding mode.78 The existing results suggest that the complexes can bind to CT DNA, although the exact mode of DNA-binding cannot be concluded only by UV spectroscopic titration studies; nevertheless, more experiments are necessary in order to further clarify the binding mode.78
| Compound | Band (ΔA/A0a, Δλb) | K b (M−1) |
|---|---|---|
| a “+” denotes hyperchromism, “−” denotes hypochromism. b “+” denotes red-shift, “−” denotes blue-shift. | ||
| Hflmq33 | 326 (−15, +2), 342 (+5, +2) | 3.53 (±0.45) × 105 |
| [Co(flmq)2(MeOH)2], 1 | 329 (+1, +2), 340 (−4, +1) | 5.76 (±0.16) × 105 |
| [Co(flmq)2(bipy)], 2 | 329 (+4, −3), 338 (−5, 0) | 4.73 (±0.11) × 105 |
| [Co(flmq)2(phen)], 3 | 327 (+4, 0), 339 (−6, 0) | 1.41 (±0.22) × 105 |
| [Co(flmq)2(bipyam)], 4 | 322 (+6, +2), 340 (−3, 0) | 7.88 (±0.12) × 105 |
The values of the DNA-binding constant (Kb) of complexes 1–4 (Table 3) were calculated from the plots [DNA]/(εA − εf) versus [DNA] (Fig. S4†) and the Wolfe–Shimer equation (eqn (S1)†).61 The Kb constants are relatively high and of the same magnitude to that of free Hflmq. The Kb constants indicate a strong binding of the complexes to CT DNA, with complex 4 bearing the highest Kb constant (=7.88(±0.12) × 105 M−1) among the compounds, and are higher than that of the classical intercalator EB (=1.23(±0.07) × 105 M−1) as calculated in our lab.79 The Kb constants of the complexes are among the highest constants of the metal–quinolone complexes reported.28
Electrochemical techniques may be used in complement when studying the interaction of complexes with DNA and useful information concerning the binding mode of the reduced and oxidized form of the complex with DNA may arise. Cyclic voltammetry is among the most common electrochemical techniques used for such studies since the shift of the electrochemical potentials of the complex in the presence of DNA may reveal their interaction mode; a positive shift occurs upon intercalation and a negative shift is a result of an electrostatic interaction between the complex and DNA.80,81
In particular, the cyclic voltammograms of complexes 1–4 (0.4 mM) in 1/2 DMSO/buffer solution were recorded in the presence of CT DNA (representatively shown for 2 in Fig. S5†), the cathodic (Epc) and anodic (Epa) potentials of the quasi-reversible redox couple Co(II)/Co(I) were found and their shifts were calculated (Table 4). The cathodic and the anodic potentials exhibited in the presence of CT DNA mainly a positive shift (ΔEp = (−6)–(+14) mV) suggesting intercalation as the most likely interaction mode between the complexes and CT DNA bases;81 a conclusion which sheds light on the findings of the spectroscopic studies and is in accordance with the viscosity experiments.
| Complex | E pc(f) a | E pc(b) b | ΔEpcc | E pa(f) a | E pa(b) b | ΔEpac |
|---|---|---|---|---|---|---|
| a E pc/a in DMSO/buffer in the absence of CT DNA (Epc/a(f)). b E pc/a in DMSO/buffer in the presence of CT DNA (Epc/a(b)). c ΔEpc/a = Epc/a(b) − Epc/a(f). | ||||||
| [Co(flmq)2(MeOH)2], 1 | −706 | −696 | +10 | −563 | −553 | +10 |
| [Co(flmq)2(bipy)], 2 | −711 | −704 | +7 | −557 | −563 | −6 |
| [Co(flmq)2(phen)], 3 | −713 | −705 | +8 | −526 | −532 | −6 |
| [Co(flmq)2(bipyam)], 4 | −717 | −703 | +14 | −530 | −530 | 0 |
The study of the DNA viscosity in the presence of a compound may provide significant information in regard to the DNA-binding mode, since the relative DNA-viscosity (η/η0) is rather sensitive to changes of the relative DNA-length (L/L0) as they are related by the equation L/L0 = (η/η0)1/3.18,43 The viscosity of a CT DNA solution (0.1 mM) was monitored in the presence of increasing amounts of complexes 1–4 (up to the value of r = 0.27, Fig. 4). The relative viscosity showed an increase upon addition of the complexes; such behaviour can be attributed to a DNA-length increase arising from the insertion of the complexes in-between the DNA bases, as a result of an intercalative binding mode between DNA and each complex. Therefore, intercalation may be considered the most likely interaction mode between DNA and complexes 1–4, as concluded by the cyclic voltammetry studies, too.
 | ||
| Fig. 4 Relative viscosity (η/η0)1/3 of CT DNA ([DNA] = 0.1 mM) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0) in the presence of complexes 1–4 at increasing amounts (r = [complex]/[DNA]). | ||
Ethidium bromide (EB = 3,8-diamino-5-ethyl-6-phenyl-phenanthridinium bromide) intercalates to CT DNA via its planar phenanthridine ring in-between adjacent base pairs of the DNA double helix; thus, enhanced fluorescence emission due to the formation of the EB–DNA compound appears. EB is a typical indicator of intercalation; the quenching of the DNA–EB fluorescence emission may occur with the co-existence of a compound acting as a DNA-intercalator equally or more strongly than EB.82 Complexes 1–4 did not exhibit any significant fluorescence emission at room temperature in solution or in the presence of CT DNA, when excited at 540 nm, and their addition to an EB-solution did not induce any quenching of the free EB fluorescence or the appearance of new peaks in the fluorescence emission spectra. Within this context, the changes observed in the fluorescence emission spectra of the EB–DNA solution in the presence of 1–4 were used to study the complexes’ EB-displacing ability from the EB–DNA complex.
The fluorescence emission spectra (λexcit = 540 nm) of pre-treated EB–CT DNA ([EB] = 20 μM, [DNA] = 26 μM) exhibited a band of significant intensity at λem,max = 592 nm as a result of EB-intercalating into DNA base pairs. This band presented an intense quenching upon addition of the complexes at increasing amounts up to an r value = 0.22 (Fig. 5(A) representatively for complex 3). The addition of complexes 1–4 into the EB–DNA solution resulted in a rather significant quenching (Fig. 5(B)) of the band at 592 nm (the final fluorescence is up to 26–36% of the initial EB–DNA fluorescence intensity in the presence of the complexes, Table 5); thus, the complexes may displace EB from the EB–DNA compound and subsequently can intercalate to CT DNA.28
 | ||
| Fig. 5 (A) Fluorescence emission spectra (λexcit = 540 nm) for EB–DNA ([EB] = 20 μM, [DNA] = 26 μM) in buffer solution in the absence and presence of increasing amounts of complex 3 (up to the r value = 0.21). The arrow shows the changes of intensity upon increasing amounts of 3. (B) Plot of EB relative fluorescence emission intensity at λem = 592 nm (%) vs. r (r = [complex]/[DNA]) (150 mM NaCl and 15 mM trisodium citrate at pH = 7.0) in the presence of complexes 1–4 (up to 25.7% of the initial EB–DNA fluorescence emission intensity for 1, 33.3% for 2, 31.0% for 3 and 36.0% for 4). | ||
| Compound | ΔI/Io (%) | K SV (M−1) |
|---|---|---|
| Hflmq33 | 55.0 | 1.19(±0.06) × 106 |
| [Co(flmq)2(MeOH)2], 1 | 74.3 | 1.27(±0.24) × 105 |
| [Co(flmq)2(bipy)], 2 | 66.7 | 1.73(±0.75) × 105 |
| [Co(flmq)2(phen)], 3 | 69.0 | 1.61(±0.49) × 105 |
| [Co(flmq)2(bipyam)], 4 | 64.0 | 6.24(±0.27) × 105 |
The Stern–Volmer plots of EB–DNA fluorescence quenching induced by complexes 1–4 (Fig. S6†) illustrate the good agreement (R = 0.99) of the quenching of EB–DNA with the linear Stern–Volmer equation (eqn (S2)†), proving, thus, that the replacement of EB from EB–DNA by each compound results in a decrease in the fluorescence intensity.33–36 The KSV constants (Table 5) are relatively high showing the tight bind of the complexes to DNA; complex 4 exhibits the highest KSV constant (=6.24(±0.27) × 105 M−1) among the complexes. The KSV constants of complexes 1–4 are similar with those reported for a series of metal complexes with flumequine and other quinolones as ligands.28
Interaction of complexes 1–4 with serum albumin
Serum albumin (SA) is responsible for the transport of ions and drugs through the bloodstream to cells and tissues and they are the most abundant proteins in plasma.63 The investigation of the interaction of biologically active compounds (such as complexes 1–4) with SA is important, since differentiated biological properties of the compound or novel transport pathways towards their targets in the body may arise.83 Within this context, the interaction of complexes 1–4 with human serum albumin (HSA) and bovine serum albumin (BSA) was studied; BSA is homologous to HSA and is the most studied SA. The solutions of the SA exhibit an intense fluorescence emission when excited at 295 nm, with λem,max = 351 nm for HSA and 343 nm for BSA due to the existence of tryptophans; a tryptophan at position 214 in HSA and two tryptophans located at position 134 and 212 for BSA.63 The flumequine compounds 1–4 exhibited an emission band with λem,max at 365 nm under the same experimental conditions, i.e. excitation at 295 nm;33–36 thus, the SA fluorescence spectra were corrected before the experimental data processing. The inner-filter effect was also taken into consideration and was calculated with eqn (S3);† it was not found to be significant and only slightly affected the measurements.64The fluorescence emission spectra of HSA and BSA exhibited in the presence of complexes 1–4 a moderate (for HSA) to significant (for BSA) quenching of the fluorescence (quenching of the initial SA fluorescence up to ∼61% and ∼75% in the presence of complex 2 for HSA and BSA, respectively, Fig. 6). The observed quenching in the fluorescence emission spectra of the SAs may be due to possible changes in the tryptophan environment of SA which are induced by changes in albumin secondary structure as a result of the binding of each complex to SA.84
 | ||
| Fig. 6 (A) Plot of % relative fluorescence intensity at λem = 351 nm (%) vs. r (r = [complex]/[HSA]) for complexes 1–4 (up to 48.5% of the initial HSA fluorescence for 1, 39.0% for 2, 64.5% for 3 and 67.1% for 4) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0). (B) Plot of % relative fluorescence intensity at λem = 343 nm (%) vs. r (r = [complex]/[BSA]) for complexes 1–4 (up to 48.1% of the initial BSA fluorescence for 1, 25.3% for 2, 33.5% for 3 and 25.0% for 4) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0). | ||
The quenching constants (kq) for the interaction of complexes 1–4 with the albumins were calculated from the corresponding Stern–Volmer plots (Fig. S7 and S8†) by the Stern–Volmer quenching equation (eqn (S4)†) and their values are given in Tables 6 and S5.† The determined values of kq suggest significant SA quenching ability and they are significantly higher than 1012 M−1 s−1 suggesting thus the existence of a static quenching mechanism.62 The kq constants of the complexes are of the same magnitude to the those of free Hflmq, with 2 and 4 exhibiting the highest kq for BSA (kq(BSA),2 = 1.67(±0.03) × 1013 M−1 s−1 and kq(BSA),4 = 1.59(±0.09) × 1013 M−1 s−1) and 2 for HSA (kq(hSA),2 = 9.00(±0.31) × 1012 M−1 s−1). The values of the kq are within the range found for a series of metal-complexes bearing flumequine33–36 and other quinolones as ligands.28
| Compound | k q(BSA) (M−1 s−1) | K (BSA) (M−1) | k q(HSA) (M−1 s−1) | K (HSA) (M−1) |
|---|---|---|---|---|
| Hflmq33 | 8.26(±0.36) × 1012 | 6.67 × 104 | 1.00(±0.17) × 1013 | 2.37 × 106 |
| [Co(flmq)2(MeOH)2], 1 | 5.22(±0.32) × 1012 | 1.57(±0.11) × 105 | 3.64(±0.25) × 1012 | 5.04(±0.26) × 105 |
| [Co(flmq)2(bipy)], 2 | 1.67(±0.03) × 1013 | 1.43(±0.07) × 105 | 9.00(±0.31) × 1012 | 2.90(±0.27) × 104 |
| [Co(flmq)2(phen)], 3 | 1.13(±0.03) × 1013 | 7.92(±0.12) × 104 | 3.17(±0.14) × 1012 | 8.18(±0.35) × 103 |
| [Co(flmq)2(bipyam)], 4 | 1.59(±0.09) × 1013 | 9.75(±0.32) × 104 | 2.65(±0.11) × 1012 | 6.03(±0.33) × 104 |
The binding constants (K) of the complexes to both the albumins were determined from the corresponding Scatchard plots (Fig. S9 and S10†) using the Scatchard equation (eqn (S6)†) and are given in Tables 6 and S5.† The K constants of all complexes 1–4 are relatively high and are of the same magnitude to those calculated for a series of metal complexes with flumequine33–36 and other quinolones as ligands.28 The complexes exhibit for BSA higher affinity than free Hflmq with complexes 1 and 2 having the highest K constants (K(BSA),1 = 1.57(±0.11) × 105 M−1 and K(BSA),2 = 1.43(±0.07) × 105 M−1), while the affinity of the complexes for HSA is lower than free Hflmq with complex 1 bearing the highest K constant among the complexes (K(HSA),4 = 5.04(±0.26) × 105 M−1).
In general, the K constants of complexes 1–4 are in the range 8.18 × 103 to 5.04 × 105 M−1 and are relatively high showing the ability of the compounds to bind to albumins and get transferred by them towards their targets cells or tissues. A comparison of the K values to the association constant of avidin with diverse ligands (K ≈ 1015 M−1, such interactions are considered as the strongest known non-covalent interaction)85 may reveal the ability of the compounds to get released from the albumins probably upon arrival at their targets.84
Biological activity of the complexes
| E. coli | X. campestris | B. subtilis | S. aureus | |||||
|---|---|---|---|---|---|---|---|---|
| MIC | IC50 | MIC | IC50 | MIC | IC50 | MIC | IC50 | |
| Hflmq | 1(3.82) | 0.56(2.14) | 8(30.53) | 2.82(10.76) | 2(7.64) | 1.47(5.61) | 16(61.06) | 7.5(28.63) |
| [Co(flmq)2(MeOH)2], 1 | 4(6.22) | 1.92(2.98) | 16(24.88) | 6.66(10.35) | 4(6.22) | 3.00(4.66) | 64(99.46) | 32.3(50.19) |
| [Co(flmq)2(bipy)], 2 | 2(2.59) | 0.87(1.13) | 8(10.36) | 4.72(6.12) | 4(5.18) | 2.52(3.27) | 32(41.45) | 16.3(21.12) |
| [Co(flmq)2(phen)], 3 | 2(2.44) | 0.99(1.21) | 8(9.78) | 5.28(6.45) | 4(4.89) | 2.57(3.14) | 64(78.23) | 27.6(33.74) |
| [Co(flmq)2(bipyam)], 4 | 2(2.60) | 0.86(1.19) | 16(20.82) | 4.97(6.47) | 2(2.60) | 1.51(1.96) | 32(41.63) | 12.9(16.78) |
| [Cu(flmq)2(H2O)2], 5 | 2(3.32) | 1.22(2.03) | 8(13.29) | 2.84(4.72) | 2(3.32) | 1.52(2.52) | >64(>106.30) | 40.2(66.78) |
| [Cu(flmq)(bipy)Cl], 6 | 2(3.63) | 1.10(1.99) | 8(14.51) | 5.73(10.39) | 4(7.25) | 2.05(3.72) | 32(58.03) | 18.4(33.37) |
| [Cu(flmq)(phen)Cl], 7 | 2(3.18) | 1.35(2.14) | 8(12.71) | 4.70(7.47) | 2(3.18) | 1.32(2.10) | 32(50.83) | 16.8(26.89) |
| [Cu(flmq)(bipyam)Cl], 8 | 2(3.65) | 1.01(1.84) | 8(14.59) | 5.09(9.28) | 4(7.29) | 1.33(2.43) | 32(58.35) | 19.2(35.01) |
| [Cu(flmq)2(py)2], 9 | 4(5.39) | 1.56(2.10) | 8(10.79) | 5.68(7.65) | 8(10.79) | 4.32(5.82) | 16(21.56) | 11.8(15.90) |
Flumequine and its cobalt(II) and copper(II) complexes present inhibitory action against all the microorganisms tested, especially against E. coli and B. subtilis, with MIC and IC50 values in the range 1–8 μg mL−1 (2.44–10.79 μM) and 0.56–3.00 μg mL−1 (1.13–5.82 μM), respectively. It is clear that the complexes are similarly or slightly more active than free Hflmq against the microorganisms tested (Table 7). Based on the concentrations expressed in molarity units, we may conclude that the antimicrobial activity of the complexes is up to three times higher than the activity of free Hflmq. The compounds are active against E. coli and B. subtilis (MIC = 2–4 μg mL−1 (2.44–10.79 μM)), while they do not seem to be very active against the most resistant S. aureus strain (MIC = 16–64 μg mL−1 (21.56–106.3 μM)).
There is no noteworthy differentiation on the activity observed for the Co(II)–flmq complexes 1–4, the Cu(II)–flmq complexes 5–9 and their corresponding Zn(II)–flmq complexes recently reported.36 Therefore, we may conclude that for such low MIC values, the nature of the metal does not play a significant role in the antimicrobial activity and the observed activity of the complexes may be mainly attributed to the presence of the quinolone ligands. Complexes 1–4 are less active than the corresponding Co(II)–enrofloxacinato complexes recently reported,18 since enrofloxacin is a second-generation quinolone, and its compounds may be more active than those of a first-generation quinolone such as flumequine.
Among the five factors responsible for the antimicrobial activity of a complex (chelate effect of ligands, nature of ligands, nuclearity, total charge, existence and nature of counterions),86 the nature of the ligands (the quinolone flumequine and the oxygen- or nitrogen-donors) and the chelate effect of the (flumequinato and nitrogen-donor) ligands seem to influence mainly the antimicrobial activity of the complexes. There are no evident differences that could be attributed to the nature of the co-ligands (bipy, phen, bipyam); the other three factors (nuclearity, total charge and existence of counterions) do not contribute to diversity of the antimicrobial activity since all complexes are mononuclear and neutral.
The determination of MIC is among the generally accepted methods used for testing the effectiveness of antibiotics on inhibiting microbial growth. On the other hand, the simultaneous cultivation and on-line analysis of the microorganisms in the presence of antimicrobial agents is used to provide more information, regarding the effect of sub-MIC concentrations on growth and the effect on bacterial population growth kinetics, than the static acquisition of single end-point determination.87–89 It was shown that flumequine at MIC and MIC/2 affects the elongation of the lag phase and the reduction of growth rate, whereas in the presence of complexes 3 and 6, a significant increase of the lag phase is observed but there was little or no effect on maximum growth rate (Fig. 7). The results indicate that the bacterial growth curve is affected differently by flumequine and by its complexes, which could be attributed to a different cell intake route as has been recently suggested.10 To our knowledge, this is the first time that this method is employed for the determination of the antibacterial activity of metal complexes.
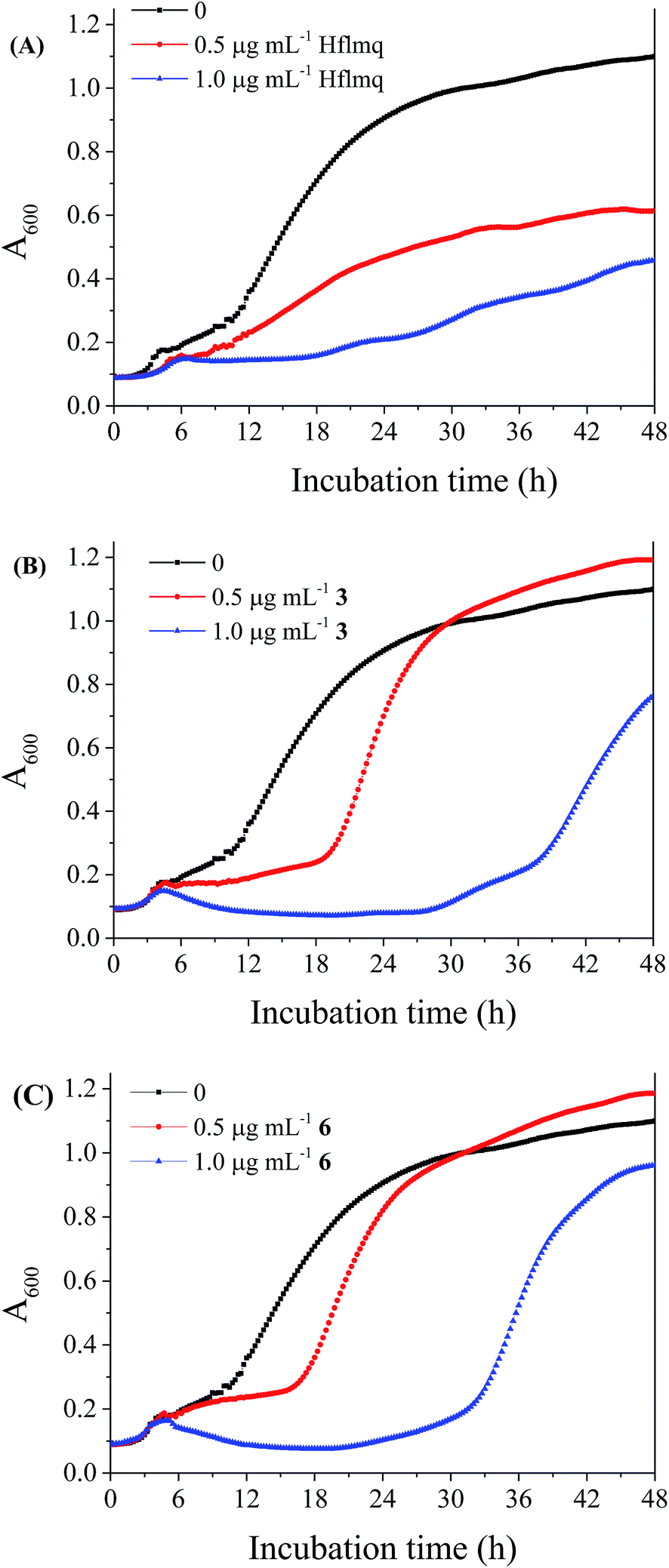 | ||
| Fig. 7 Growth of E. coli in the presence of (A) Hflmq, (B) [Co(flmq)2(phen)], 3 and (C) [Cu(flmq)(bipy)Cl], 6 at three different concentrations (no compound, 0.5 μg mL−1 of compound and 1 μg mL−1 of compound). All cultures were grown in 8% v/v DMSO. | ||
In conclusion, the best inhibition is provided by the complexes against E. coli and B. subtilis (MIC = 2–4 μg mL−1), while S. aureus rather exhibits resistance to the activity of the compounds. Despite the fact that the complexes are not significantly more active than free flumequine, their IC50 and MIC values are low and the complexes might be considered promising for their potency as antibacterial agents.
| Compound | A2780 | A549 |
|---|---|---|
| Hflmq | >100 | >200 |
| phen | 3.7 ± 1.5 | 22 ± 4 |
| [Co(flmq)(phen)Cl], 3 | >50 | >50 |
| [Cu(flmq)(phen)Cl], 7 | 0.26 ± 0.07 | 3.2 ± 0.4 |
| [Zn(flmq)(phen)Cl], 10 | 2.40 ± 0.03 | 3.6 ± 0.7 |
| [Ni(flmq)2(phen)], 11 | 13 ± 2 | >100 |
| Cisplatin | 1.5 ± 0.7 | 11 ± 1.1 |
While Hflmq does not appear to be toxic in both the tested cell lines, phen has a low IC50 in A2780 and moderate effect in A549 cells. The cobalt complex 3 is poorly cytotoxic with IC50 values above 50 μM. Interestingly, although Hflmq has similar antibacterial properties to complex 7 in most of the tested bacterial strains, complex 7 appears to be very potent against both cancer cell lines, with a low sub-micromolar IC50 in A2780. Moreover, the zinc complex 10 displays similar toxicity for both cell lines, in the same range as complex 7 in A549 cells. On the other hand, the nickel complex 11 has only moderate toxicity on A2780 and practically no toxicity on A549 cells. The fact that the ligand phen also shows a high toxicity against the two cell lines may also be accounting for the observed antiproliferative effect of the related complexes. Nonetheless, it is noteworthy that complex 7 is ca. 10-fold more toxic than phen. On the other hand, Ni2+ complex 11 is less toxic than phen. Remarkably, the Zn2+ complex 10, at variance with the other complexes and both ligands, does not display selectivity against A2780 cells and has a similar IC50 in both tested cell lines, in the low μM range.
It is worth noting that among the four complexes studied for their antiproliferative activity, complex 7 exhibited the highest affinity for the albumins while the DNA-binding constant of complex 10 was two to five times higher than that of complexes 3, 7 and 11 (Table S6†). Therefore, we may conclude that the most cytotoxic complexes 7 and 10 presented the highest binding affinity for albumins and CT DNA, respectively.
Conclusions
The synthesis and characterization of the cobalt(II) complexes with the first-generation quinolone flumequine in the absence or presence of the α-diimines 2,2′-bipyridine, 1,10-phenanthroline or 2,2′-bipyridylamine was achieved. In the resultant complexes, the deprotonated flumequine ligands are bidentately coordinated to cobalt via the pyridone and a carboxylato oxygen. The crystal structures of the complexes [Co(flmq)2(bipy)]·2H2O, 2·2H2O, [Co(flmq)2(phen)]·1.6MeOH·0.4H2O, 3·1.6MeOH·0.4H2O and [Co(flmq)2(bipyam)]·H2O, 4·H2O were determined by X-ray crystallography. The geometry around Co(II) is distorted octahedral and the arrangement of the oxygen atoms around the cobalt is similar in complexes 2 and 3 (cis carboxylato oxygens and trans pyridone oxygens), while in complex 4 the arrangement of the oxygens is inverted. Such a difference in the arrangement of the coordinated oxygen atoms around the metal was also observed in the Ni(II) and Zn(II) complexes bearing flumequinato ligands.The interaction of the Co(II)–flumequine complexes with CT DNA was monitored by UV spectroscopy, viscosity measurements, cyclic voltammetry and competitive studies with EB. According to all techniques used, intercalation is the most likely interaction mode of the complexes to CT DNA. Complex [Co(flmq)2(bipyam)], 4, exhibits the highest DNA-binding constant (Kb = 7.88(±0.12) × 105 M−1), among the Co(II)–flumequine complexes.
The affinity of complexes 1–4 with bovine or human serum albumins was investigated by fluorescence emission spectroscopy; the complexes exhibit tight binding affinity to BSA and HSA with relatively high SA-binding constants (K = 8.18 × 103 to 5.04 × 105 M−1). The obtained K constants are indicative of the binding of the complexes to the albumins and their potential for transportation and release when arriving at their targets.
The antimicrobial activity of the Co(II)–flumequine and the previously reported Cu(II)–flumequine complexes was evaluated by the MIC and the IC50 values and was comparable to that of free Hflmq against the four bacteria tested. The best inhibition of complexes 1–9 is against E. coli; on the other hand, the compounds are significantly less active against the most resistant microorganism S. aureus in the range of the concentrations tested.
Finally, the new cobalt complex 3 showed poor antiproliferative effects in vitro in cancer cells, while the copper and zinc analogues are promising cytotoxic agents. The present results on the interaction of the compounds with DNA or SA support the preliminary idea that the magnitude of binding to biomolecules such DNA or SA may be related to the cytotoxicity. However, the number of present compounds is limited so it is not possible to form concrete conclusions in regard to a possible structure–activity relationship between cytotoxicity and binding to biomolecules. Interestingly, the diversity in the observed anticancer effects may be due to different accumulation pathways and/or mode of actions for the various metal compounds. Furthermore, it should be noted that other biological targets should be considered, such as for example damage of intracellular zinc finger proteins, as observed for Co2+ ions in previous studies.90
Abbreviations
| A2780 | Human ovarian carcinoma cells |
| A549 | Human lung carcinoma cells |
| B. subtilis | Bacillus subtilis ATCC 6633 |
| bipy | 2,2′-Bipyridine |
| bipyam | 2,2′-Bipyridylamine |
| BSA | Bovine serum albumin |
| CT | Calf-thymus |
| DMF | N,N-Dimethylformamide |
| E. coli | Escherichia coli NCTC 29212 |
| EB | Ethidium bromide, 3,8-diamino-5-ethyl-6-phenyl-phenanthridinium bromide |
| FBS | Fetal bovine serum |
| Hflmq | 6,7-Dihydro-9-fluoro-5-methyl-1-oxo-1H,5H-[ij]quinolizine-2-carboxylic acid |
| HSA | Human serum albumin |
| IC50 | Half-minimum inhibitory concentration |
| K | SA-binding constant |
| K b | DNA-binding constant |
| k q | SA-quenching constant |
| K SV | Stern–Volmer constant |
| MIC | Minimum inhibitory concentration |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| phen | 1,10-Phenanthroline |
| py | Pyridine |
| r | [Complex]/[DNA] |
| r′ | [DNA]/[complex] |
| s | Strong |
| S. aureus | Staphylococcus aureus ATCC 6538 |
| SA | Serum albumin |
| sh | Shoulder |
| vs | Very strong |
| X. campestris | Xanthomonas campestris ATCC 1395 |
| Δν(CO2) | ν asym(CO2) − νsym(CO2) |
Acknowledgements
This research has been co-financed by European Social Fund (ESF) and Greek national funds (National Strategic Reference Framework (NSRF)): Archimides III. Financial support from the Slovenian Research Agency (ARRS) through project P1-0175 is gratefully acknowledged. We thank the EN-FIST Centre of Excellence, Ljubljana, Slovenia, for using SuperNova diffractometer. This project was also supported by EU COST Action CM1105: “Functional metal complexes that bind to biomolecules”.References
- The Quinolones, ed. V. T. Andriole, Academic Press, 2000 Search PubMed.
- D. E. King, R. Malone and S. H. Lilley, Am. Fam. Physician, 2000, 61, 2741–2748 CAS.
- I. Turel, Coord. Chem. Rev., 2002, 232, 27–47 CrossRef CAS and references cited therein.
- D. C. Hooper and E. Rubinstein, Quinolone Antimicrobial Agents, ASM Press, Washington, DC, 3rd edn, 2003 Search PubMed.
- J. Tuma, W. H. Connors, D. H. Stitelman and C. Richert, J. Am. Chem. Soc., 2002, 124, 4236–4246 CrossRef CAS PubMed.
- J. H. Morais Cabral, A. P. Jackson, C. V. Smith, N. Shikotra, A. Maxwell and R. C. Liddington, Nature, 1997, 388, 903–906 CrossRef CAS PubMed.
- N. Ahmed, M. Dawson, C. Smith and E. Wood, Biology of Disease, Taylor & Francis, 2007, ch. 3, pp. 41–71 Search PubMed.
- K. Drlica, A. Mustaev, T. R. Towle, G. Luan, R. J. Kerns and J. M. Berger, ACS Chem. Biol., 2014, 9, 2895–2904 CrossRef CAS PubMed.
- K. J. Aldred, S. A. McPherson, C. L. Turnbough Jr, R. J. Kerns and N. Osheroff, Nucleic Acids Res., 2013, 41, 4628–4639 CrossRef CAS PubMed.
- M. J. Feio, I. Sousa, M. Ferreira, L. Cunha-Silva, R. G. Saraiva, C. Queirós, J. G. Alexandre, V. Claro, A. Mendes, R. Ortiz, S. Lopes, A. L. Amaral, J. Lino, P. Fernandes, A. J. Silva, L. Moutinho, B. de Castro, E. Pereira, L. Perello and P. Gameiro, J. Inorg. Biochem., 2014, 138, 129–143 CrossRef CAS PubMed.
- K. Sandstrom, S. Warmlander, M. Leijon and A. Graslund, Biochem. Biophys. Res. Commun., 2003, 304, 55–59 CrossRef CAS PubMed.
- N. Jimenez-Garrido, L. Perello, R. Ortiz, G. Alzuet, M. Gonzalez-Alvarez, E. Canton, M. Liu-Gonzalez, S. Garcia-Granda and M. Perez-Priede, J. Inorg. Biochem., 2005, 99, 677–689 CrossRef CAS PubMed.
- P. Drevensek, T. Zupancic, B. Pihlar, R. Jerala, U. Kolitsch, A. Plaper and I. Turel, J. Inorg. Biochem., 2005, 99, 432–442 CrossRef CAS PubMed.
- J. A. Wiles, Q. Wang, E. Lucien, A. Hashimoto, Y. Song, J. Cheng, C. W. Marlor, Y. Ou, S. D. Podos, J. A. Thanassi, C. Thoma, M. Deshpande, M. J. Pucci and B. J. Bradbury, Bioorg. Med. Chem. Lett., 2006, 16, 1272–1276 CrossRef CAS PubMed.
- V. Uivarosi, Molecules, 2013, 18, 11153–11197 CrossRef CAS PubMed.
- I. Turel, A. Golobic, A. Klavzar, B. Pihlar, P. Buglyo, E. Tolis, D. Rehder and K. Sepcic, J. Inorg. Biochem., 2003, 95, 199–207 CrossRef CAS PubMed.
- M. P. Lopez-Gresa, R. Ortiz, L. Perello, J. Latorre, M. Liu-Gonzalez, S. Garcia-Granda, M. Perez-Priede and E. Canton, J. Inorg. Biochem., 2002, 92, 65–74 CrossRef CAS PubMed.
- C. Protogeraki, E. G. Andreadou, F. Perdih, I. Turel, A. A. Pantazaki and G. Psomas, Eur. J. Med. Chem., 2014, 86, 189–201 CrossRef CAS PubMed.
- Y. Xia, Z. Y. Yang, P. Xia, K. F. Bastow, Y. Tachibana, S. C. Kuo, E. Hamel, T. Hackl and K. H. Lee, J. Med. Chem., 1998, 41, 1155–1162 CrossRef CAS PubMed.
- J. Kljun, A. K. Bytzek, W. Kandioller, C. Bartel, M. A. Jakupec, C. G. Hartinger, B. K. Keppler and I. Turel, Organometallics, 2011, 30, 2506–2512 CrossRef CAS PubMed.
- I. Turel, J. Kljun, F. Perdih, E. Morozova, V. Bakulev, N. Kasyanenko, J. A. W. Byl and N. Osheroff, Inorg. Chem., 2010, 49, 10750–10752 CrossRef CAS PubMed.
- E. K. Efthimiadou, H. Thomadaki, Y. Sanakis, C. P. Raptopoulou, N. Katsaros, A. Scorilas, A. Karaliota and G. Psomas, J. Inorg. Biochem., 2007, 101, 64–73 CrossRef CAS PubMed.
- M. E. Katsarou, E. K. Efthimiadou, G. Psomas, A. Karaliota and D. Vourloumis, J. Med. Chem., 2008, 51, 470–478 CrossRef CAS PubMed.
- J. Kljun, I. Bratsos, E. Alessio, G. Psomas, U. Repnik, M. Butinar, B. Turk and I. Turel, Inorg. Chem., 2013, 52, 9039–9052 CrossRef CAS PubMed.
- L. Wang, Y. Wu, Y. Tan, X. Fei, Y. Deng, H. Cao, B. Chen, H. Wang, J. Magdalou and L. Chen, J. Appl. Toxicol., 2014, 34, 870–877 CrossRef CAS PubMed.
- L. R. Gouvea, L. S. Garcia, D. R. Lachter, P. R. Nunes, F. de Castro Pereira, E. P. Silveira-Lacerda, S. R. W. Louro, P. J. S. Barbeira and L. R. Teixeira, Eur. J. Med. Chem., 2012, 55, 67–73 CrossRef CAS PubMed.
- B. Duff, V. R. Thangella, B. S. Creaven, M. Walsh and D. A. Egan, Eur. J. Pharmacol., 2012, 689, 45–55 CrossRef CAS PubMed.
- G. Psomas and D. P. Kessissoglou, Dalton Trans., 2013, 42, 6252–6276 RSC.
- D. Greenwood, Antimicrob. Agents Chemother., 1978, 13, 479–483 CrossRef CAS PubMed.
- A. Ruiz-Garcia, M. Bermejo, V. Merino, G. Sanchez-Castano, J. Freixas and T. M. Garrigues, Eur. J. Pharm. Biopharm., 1999, 48, 253–258 CrossRef CAS PubMed.
- C. R. Steer, C. L. Huby, A. P. Ball, R. J. Dickinson, S. Pickens, E. T. Wallace, A. M. M. Wilson and J. A. Gray, J. Antimicrob. Chemother., 1981, 7, 643–648 CrossRef CAS PubMed.
- J. M. Delmas, A. M. Chapel and P. Sanders, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 712, 263–268 CrossRef CAS.
- K. C. Skyrianou, F. Perdih, I. Turel, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2010, 104, 740–749 CrossRef CAS PubMed.
- E. Chalkidou, F. Perdih, I. Turel, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2012, 113, 55–65 CrossRef CAS PubMed.
- A. Tarushi, J. Kljun, I. Turel, A. A. Pantazaki, G. Psomas and D. P. Kessissoglou, New J. Chem., 2013, 37, 342–355 RSC.
- A. Tarushi, K. Lafazanis, J. Kljun, I. Turel, A. A. Pantazaki, G. Psomas and D. P. Kessissoglou, J. Inorg. Biochem., 2013, 121, 53–65 CrossRef CAS PubMed.
- P. V. Bernhardt and G. A. Lawrance, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, 2003, vol. 6, ch. 1, pp. 1–45 Search PubMed.
- P. J. Sadler, Adv. Inorg. Chem., 1991, 36, 1–48 CrossRef CAS.
- M. D. Hall, T. W. Failes, N. Yamamoto and T. W. Hambley, Dalton Trans., 2007, 3983–3990 RSC.
- N. J. Farrer and P. J. Sadler, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley CH, Weinheim, Germany, 2011, ch. 1, p. 16 Search PubMed.
- D. U. Miodragovic, G. A. Bogdanovic, Z. M. Miodragovic, M. D. Radulovic, S. B. Novakovic, G. N. Kaludjerovic and H. Kozlowski, J. Inorg. Biochem., 2006, 100, 1568–1574 CrossRef CAS PubMed.
- K. Nomiya, A. Yoshizawa, K. Tsukagoshi, N. C. Kasuga, S. Hirakawa and J. Watanabe, J. Inorg. Biochem., 2004, 98, 46–60 CrossRef CAS PubMed.
- E. P. Irgi, G. D. Geromichalos, S. Balala, J. Kljun, S. Kalogiannis, A. Papadopoulos, I. Turel and G. Psomas, RSC Adv., 2015, 5, 36353–36367 RSC.
- J. Lv, T. Liu, S. Cai, X. Wang, L. Liu and Y. Wang, J. Inorg. Biochem., 2006, 100, 1888–1896 CrossRef CAS PubMed.
- Z. Weiqun, Y. Wen, X. Liqun and C. Xianchen, J. Inorg. Biochem., 2005, 99, 1314–1319 CrossRef PubMed.
- F. Dimiza, A. N. Papadopoulos, V. Tangoulis, V. Psycharis, C. P. Raptopoulou, D. P. Kessissoglou and G. Psomas, Dalton Trans., 2010, 39, 4517–4528 RSC.
- F. Dimiza, A. N. Papadopoulos, V. Tangoulis, V. Psycharis, C. P. Raptopoulou, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2012, 107, 54–64 CrossRef CAS PubMed.
- H. Lopez-Sandoval, M. E. Londono-Lemos, R. Garza-Velasco, I. Poblano-Melendez, P. Granada-Macias, I. Gracia-Mora and N. Barba-Behrens, J. Inorg. Biochem., 2008, 102, 1267–1276 CrossRef CAS PubMed.
- R. Eshkourfu, B. Cobeljic, M. Vujcic, I. Turel, A. Pevec, K. Sepcic, M. Zec, S. Radulovic, T. Srdic-Radic, D. Mitic, K. Andjelkovic and D. Sladic, J. Inorg. Biochem., 2011, 105, 1196–1203 CrossRef CAS PubMed.
- A. Bottcher, T. Takeuchi, K. I. Hardcastle, T. J. Meade and H. B. Gray, Inorg. Chem., 1997, 36, 2498–2504 CrossRef.
- T. Takeuchi, A. Bottcher, C. M. Quezada, T. J. Meade and H. B. Gray, Bioorg. Med. Chem., 1999, 7, 815–819 CrossRef CAS PubMed.
- N. Hadjiliadis and E. Sletten, Metal complex–DNA interactions, John Wiley & Sons, Inc., Hoboken, NJ, 2009 and references therein Search PubMed.
- N. Jimenez-Garrido, L. Perello, R. Ortiz, G. Alzuet, M. Gonzalez-Alvarez, E. Canton, M. Liu-Gonzalez, S. Garcia-Granda and M. Perez-Priede, J. Inorg. Biochem., 2005, 99, 677–689 CrossRef CAS PubMed.
- J. He, D. Xiao, H. Chen, D. Sun, S. Yan, X. Wang, Z. Ye, Q. Luo and E. Wang, J. Solid State Chem., 2013, 198, 279–288 CrossRef CAS.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–211 CrossRef CAS.
- M. F. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- Agilent, CrysAlis PRO, Agilent Technologies, Yarnton, Oxfordshire, England, 2011 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Wolfe, G. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- G. Zhao, H. Lin, S. Zhu, H. Sun and Y. Chen, J. Inorg. Biochem., 1998, 70, 219–226 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- L. Stella, A. L. Capodilupo and M. Bietti, Chem. Commun., 2008, 4744–4746 RSC.
- Y. Wang, H. Zhang, G. Zhang, W. Tao and S. Tang, J. Lumin., 2007, 126, 211–218 CrossRef CAS.
- J. M. Andrews, J. Antimicrob. Chemother., 2001, 48(S1), 5–16 CrossRef CAS PubMed.
- J. Wang, H. Liu, J. Zhao, H. Gao, L. Zhou, Z. Liu, Y. Chen and P. Sui, Molecules, 2010, 15, 5807–5817 CrossRef CAS PubMed.
- V. J. Schacht, L. V. Neumann, S. K. Sandhi, L. Chen, T. Henning, P. J. Klar, K. Theophel, S. Schnell and M. Bunge, J. Appl. Microbiol., 2013, 114, 25–35 CrossRef CAS PubMed.
- E. Likotrafiti, K. M. Tuohy, G. R. Gibson and R. A. Rastall, Anaerobe, 2014, 27, 50–55 CrossRef CAS PubMed.
- C. Bresson, C. Darolles, A. Carmona, C. Gautier, N. Sage, S. Roudeau, R. Ortega, E. Ansoborlo and V. Malard, Metallomics, 2013, 5, 133–143 RSC.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, Wiley, New Jersey, 6th edn, 2009 Search PubMed.
- B. A. Frenz and J. A. Ibers, Inorg. Chem., 1972, 11, 1109–1116 CrossRef CAS.
- A. Grirrane, A. Pastor, A. Ienco, C. Mealli and A. Galindo, J. Chem. Soc., Dalton Trans., 2002, 3771–3777 RSC.
- M. M. Aleksic and V. Kapetanovic, Acta Chim. Slov., 2014, 61, 555–573 CAS.
- E. C. Long and J. K. Barton, Acc. Chem. Res., 1990, 23, 271–273 CrossRef CAS.
- Q. Zhang, J. Liu, H. Chao, G. Xue and L. Ji, J. Inorg. Biochem., 2001, 83, 49–55 CrossRef CAS PubMed.
- A. Tarushi, E. Polatoglou, J. Kljun, I. Turel, G. Psomas and D. P. Kessissoglou, Dalton Trans., 2011, 40, 9461–9473 RSC.
- G. Pratviel, J. Bernadou and B. Meunier, Adv. Inorg. Chem., 1998, 45, 251–262 CrossRef CAS.
- A. Dimitrakopoulou, C. Dendrinou-Samara, A. A. Pantazaki, M. Alexiou, E. Nordlander and D. P. Kessissoglou, J. Inorg. Biochem., 2008, 102, 618–628 CrossRef CAS PubMed.
- G. Psomas, J. Inorg. Biochem., 2008, 102, 1798–1811 CrossRef CAS PubMed.
- M. T. Carter, M. Rodriguez and A. J. Bard, J. Am. Chem. Soc., 1989, 111, 8901–8911 CrossRef CAS.
- W. D. Wilson, L. Ratmeyer, M. Zhao, L. Strekowski and D. Boykin, Biochemistry, 1993, 32, 4098–4104 CrossRef CAS PubMed.
- C. Tan, J. Liu, H. Li, W. Zheng, S. Shi, L. Chen and L. Ji, J. Inorg. Biochem., 2008, 102, 347–358 CrossRef CAS PubMed.
- V. Rajendiran, R. Karthik, M. Palaniandavar, H. Stoeckli-Evans, V. S. Periasamy, M. A. Akbarsha, B. S. Srinag and H. Krishnamurthy, Inorg. Chem., 2007, 46, 8208–8221 CrossRef CAS PubMed.
- O. H. Laitinen, V. P. Hytonen, H. R. Nordlund and M. S. Kulomaa, Cell. Mol. Life Sci., 2006, 63, 2992–3017 CrossRef CAS PubMed.
- A. D. Russell, in Disinfection, Sterilization and Preservation, ed. S. S. Block, Lippincott Williams & Wilkins, Philadelphia, 5th edn, 2001, pp. 31–56 Search PubMed.
- K. Theophel, V. J. Schacht, M. Schlüter, S. Schnell, C.-S. Stingu, R. Schaumann and M. Bunge, Front. Microbiol., 2014, 5, 544 Search PubMed.
- S. E. Walsh, J.-Y. Maillard, A. D. Russell, C. E. Catrenich, D. L. Charbonneau and R. G. Bartolo, J. Appl. Microbiol., 2003, 94, 240–247 CrossRef CAS PubMed.
- M. Gomez Escalada, A. D. Russell, J.-Y. Maillard and D. Ochs, Lett. Appl. Microbiol., 2005, 41, 476–481 CrossRef CAS PubMed.
- E. Kopera, T. Schwerdtle, A. Hartwig and W. Bal, Chem. Res. Toxicol., 2004, 17, 1452–1458 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1430329–1430331. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra25776j |
| This journal is © The Royal Society of Chemistry 2016 |