
Stereocomplex poly(lactic acid) nanoparticles crystallized through nanoporous membranes and application as nucleating agent†
Abstract
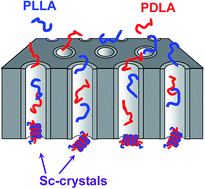
Stereocomplex crystallization of poly(L-lactic acid) (PLLA) and poly(D-lactic acid) (PDLA) was performed by flowing their blended solution through nano-channels of porous membranes. The spacing restriction within the nano-channels induces contact between the PLLA and PDLA chains. The stereocomplex crystalline structure of the obtained material was confirmed by differential scanning calorimetry and X-ray measurements. Morphological observation by scanning-electron microscopy indicated that the characteristic nano-particles with a diameter corresponding to the channel width aggregate with each other. Such nano-particles play the role of nucleating agent for matrix crystallization when they are blended with pure PLLA. The non-isothermal crystallization temperature on cooling from the melt of this blend is higher than that of pure PLLA matrix. Correspondingly, the shorter time is required for the isothermal crystallization of this blend.
 Please wait while we load your content...
Please wait while we load your content...

