
Expanding the aqueous-based redox-facilitated self-polymerization chemistry of catecholamines to 5,6-dihydroxy-1H-benzimidazole and its 2-substituted derivatives†
Ka Wai
Fan
a,
Matthew B.
Peterson
b,
Peter
Ellersdorfer
a and
Anthony M.
Granville
*a
aCentre for Advanced Macromolecular Design, School of Chemical Engineering, The University of New South Wales, Kensington, NSW 2052, Australia. E-mail: agranville999@gmail.com
bSchool of Chemistry, The University of New South Wales, Kensington, NSW 2052, Australia
First published on 1st March 2016
Abstract
Aqueous-base redox-facilitated self-polymerization can be performed with 5,6-dihydroxy-1H-benzimidazole (DHBI) to generate polymeric material that is analogous to poly(dopamine) (PDA), proving the possibility to expand the catecholamine-exclusive chemistry to N-heterocyclic catechol derivatives. DHBI underwent similar reaction pathways as dopamine to self-polymerize into the lightly cross-linked, π-conjugated poly(5,6-dihydroxy-1H-benzimidazole) (PDHBI). However, it was observed that the polymerization of DHBI proceeded faster than dopamine, and can be further enhanced under UV-stimulation, similar to dopamine polymerization. When coated on various substrates, the PDHBI coatings were capable of promoting surface wettability similar to PDA, but exhibited lower thermal stability due to a reduced cross-link density. Copolymerization compatibility between DHBI and dopamine was demonstrated, and it was possible to enhance the thermal stability of PDHBI by incorporating dopamine as a comonomer/cross-linker. Despite the high level of similarity between the two polymers, PDHBI possesses the imidazole moieties as unique features. Because of the versatile chemistry of o-benzenediamine employed for the monomer synthesis, DHBI-based monomers with specific functionality at the 2-carbon position of the imidazole ring can be prepared by choosing a desirable carboxylic acid. Two 2-substituted derivatives of DHBI were synthesized to demonstrate the ability to intrinsically modify the properties of PDHBI-based polymeric materials in terms of solubility, structure, and thermal stability.
Introduction
Since the seminal work of Lee et al.1 on the self-polymerization of dopamine, research in this area has increased dramatically. Poly(dopamine) (PDA) has been explored in: (i) the development of biocompatible2–4 and biodegradable5 drug delivery,6–8 (ii) in vivo near-infrared photothermal cancer therapy,9 (iii) electron-transfer enhancement for Li-ion batteries10,11 and artificial photosynthesis,12 (iv) facile surface modification,13,14 (v) membrane technologies for water15 and gas16 separation, and (vi) microfluidic systems.17 Although there is still conjecture as to the exact mechanism of the self-polymerization,18,19 it has been shown that the quinone form of the cyclized dopamine intermediate, 5,6-dihydroxyindole (DHIn), is the reactive species during the polymerization.20,21 As the reaction proceeds, DHIn molecules are linked through the 4-, 7- and, to a lesser degree, 2-carbon positions to form a substantially cross-linked polymeric material.Over the years, the self-polymerization technique has been applied mainly to catecholamines such as dopamine and its derivatives,22–25 norepinephrine,26,27 and L-3,4-dihydroxy-phenylalanine (L-DOPA).28,29 Recently, the range of monomers have been expanded to polyphenols and pyrogallol 2-amino-ethane,30 as well as to derivatives of DHIn with different substituents at the C-3 carbon.31,32 The use of monomers other than dopamine has the benefit of furthering the chemical versatility of the PDA-based structure. We took the structure of DHIn as inspiration and realized the potential to broaden the development of self-polymerization chemistry by substituting dopamine/DHIn with other heterocyclic catechol derivatives which are also capable of forming an activated quinone structure under the same reaction conditions. The concept grew from previous work incorporating 5-hydroxy-1H-indazole and, more recently, 5,6-dihydroxy-1H-indazole into PDA,33,34 which spawned the work reported here on the synthetic catecholic benzimidazole – 5,6-dihydroxy-1H-benzimidazole (DHBI). The successful self-polymerization of DHBI is of substantial interest to us, as the resulting polymer, poly(5,6-dihydroxy-1H-benzimidazole) (PDHBI) has the advantage of being synthesized through an aqueous-based process same as PDA while having imidazole moieties instead of pyrrole moieties within the structure. The presence of the imidazole moieties, which can potentially serve as coordination sites for ligating metals and drugs,35,36 and also binding and release site for nitric oxide,26,37,38 sparks biomedical significance for PDHBI. In addition, as indicated by Scheme 1, PDHBI shares some structural similarities with poly[2,2′-(m-phenylene)-5,5′-bisbenzimidazole], which is also known as poly(benzimidazole) (PBI).39 The outstanding mechanical, chemical and thermal stabilities, especially at elevated temperatures,40–45 of PBI makes it a desired material in several industry-orientated applications, including membrane operations,40,41 proton-conductive membranes for fuel cells,42,43 and polymer-based adhesive.44,45 Despite its industrial significance, the synthesis of PBI and its more recently improved variants is accompanied with harsh reaction conditions and often requires an inert environment.41–44 The self-polymerization of DHBI provides an alternative route to prepare materials with structure and, perhaps, performance comparable to PBIs at ambient, aqueous-based conditions. Since DHBI was synthesized based on the reaction between o-benzenediamine and carboxylic acids,46–49 this particular branch of chemistry can be used as a versatile synthesis platform to formulate a series of DHBI-based monomers with tailored functionality at the 2-carbon position without impairing the self-polymerizing ability (Scheme 2).
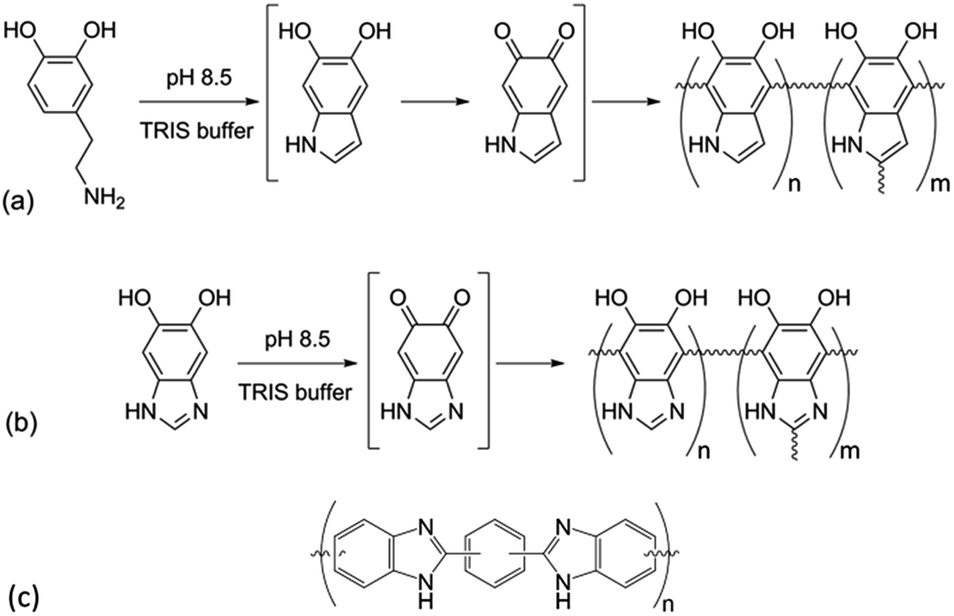 | ||
| Scheme 1 Self-polymerization of dopamine (a) and 5,6-dihydroxy-1H-benzimidazole (b), and the general structure of the industrially relevant poly(benzimidazole) (c). | ||
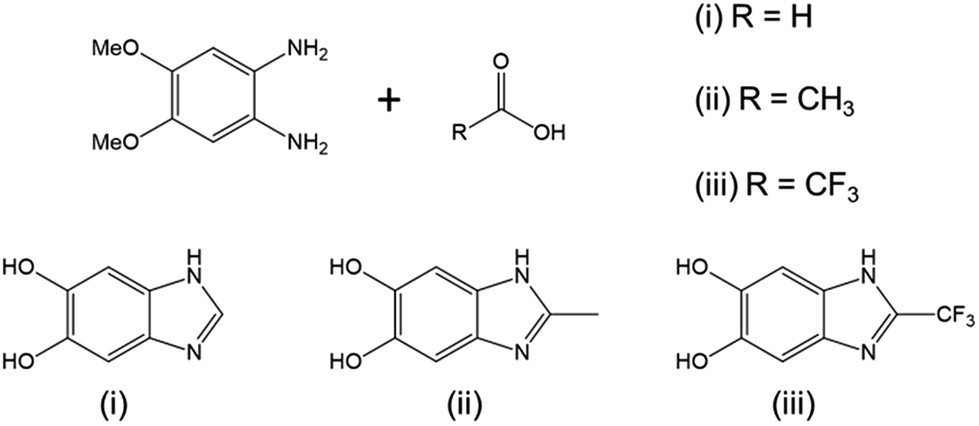 | ||
| Scheme 2 Reaction of 1,2-diamino-4,5-dimethoxybenzene with different carboxylic acids to yield DHBI carrying specific functionality at the 2-carbon position. | ||
Herein we demonstrate the capability of DHBI to self-polymerize as the sole monomer to prepare the lightly cross-linked, π-conjugated homopolymer, PDHBI, under ambient conditions in aqueous environment. It was observed that the self-polymerization of DHBI shared high level of similarities to that of dopamine in terms of the redox reaction mechanism and also the possibility to be stimulated by UV irradiation.50 The reduced cross-link density of the PDHBI structure allowed us to perform solution-based analyses on the polymer to gain further understanding of the self-polymerization and information about its polymeric nature, which would not be possible with PDA because of its poor solubility in most solvents.
Apart from studying the self-polymerization of DHBI, the present work also reveal the potential to manipulate the properties of PDHBI. To this end, we successfully copolymerized an equimolar mixture of DHBI and dopamine which not only demonstrated the compatibility of the self-polymerization system, but also showed the ability to reinforce the structure of PDHBI by increasing the degree of cross-linking with dopamine. Furthermore, we successfully synthesized two 2-substituted derivatives of DHBI – 2-methyl-5,6-dihydroxy-1H-benzimidazole (2-Me-DHBI) and 2-trifluoromethyl-5,6-dihydroxy-1H-benzimidazole (2-CF3-DHBI) – as well as preparing the respective homopolymers. These 2-substituted DHBI-based polymers were used to exemplify the potential of tailoring, for example, the structure, solubility, and thermal properties by selectively choosing the functionality at the 2-carbon position of DHBI, which in turn emphasized on the versatility of reaction between o-benzenediamine and carboxylic acids as a versatile synthesis platform.
Experimental
Materials
All reagents and solvents were used as received without further purification unless specified otherwise. Dopamine hydrochloride (99%), 1,2-dimethoxybenzene (99%), hydrazine monohydrate (98%), palladium/activated carbon catalyst (Pd/C, 10 wt%), Celite® 535, and tris(hydroxymethyl)aminomethane (TRIS) buffer (≥99.8%) were purchased from Sigma-Aldrich. Aerosil® OX 50 fumed silica particles were purchased from Evonik. Nitric acid (70%), hydrochloric acid (32%), dichloromethane (99.5%), formic acid (99%), glacial acetic acid, ethanol (95%), hydrobromic acid (48%), and pH-sticks (0–14) were purchased from Ajax Finechem Pty Ltd. Decolorizing charcoal was purchased from Ajax Chemicals. Trifluoroacetic acid (≥99%) was purchased from Merck KGaA. Deionized water was obtained from a Millipore water purification unit. The PET film, stainless steel plate and Millipore filter sheet (PVDF-based) were available resources in the laboratory and used in the coating experiment without any pretreatment.Synthesis of 1,2-dimethoxy-4,5-dinitrobenzene
The procedure was adopted from Bo et al.51 1,2-Dimethoxy-benzene (10 mL, 78.2 mmol) was added to concentrated nitric acid (65%, ca. 100 mL) dropwise under vigorous stirring over an ice-water bath. The reaction mixture was brought to room temperature, which was then heated for 2 h at 80 °C [CAUTION! nitrogen dioxide is released during the reaction]. Upon completion, the clear yellow reaction mixture was allowed to stand at room temperature and then chilled in an ice-water bath. The precipitate formed was filtered, washed with deionized water until the filtrate was neutral, and then recrystallized from hot ethanol with quantitative yield: δH (300 MHz; CDCl3), 4.01 (6H, s, OMe), 7.33 (2H, s, Ar H) ppm. δC (75 MHz; CDCl3), 57.19 (C–O), 107.1 (C-3, C-6), 136.8 (C-4, C-5), 152.0 (C-1, C-2) ppm.Synthesis of 1,2-diamino-4,5-dimethoxybenzene
The procedure was adopted from Li et al. with some modification.52 1,2-Dimethoxy-4,5-dinitrobenzene (1.0 g, 4.38 mmol) and Pd/C catalyst were dispersed in ethanol (45 mL). Hydrazine monohydrate (2.13 mL, 43.8 mmol) was added dropwise to the mixture over an ice-water bath. The mixture was refluxed for 30 min at 80 °C. Upon completion, the mixture was filtered through Celite® and the residual material was rinse with small portions of hot ethanol. The filtrate was collected and the off-white, crystalline product was obtained by removing the solvent. The product was dried and stored under nitrogen (96% yields): δH (300 MHz; CDCl3), 3.72 (6H, s, OMe), 6.31 (2H, s, Ar H) ppm. δC (75 MHz; CDCl3), 56.62 (C–O), 103.7 (C-3, C-6), 127.7 (C-1, C-2), 142.9 (C-4, C-5) ppm.Purification using decolorizing charcoal
The reaction between the synthesized o-benzenediamine and the respective carboxylic acid results in the formation of intensely colored side products, which can be removed by using decolorizing charcoal. The crude product was first dissolved in hot ethanol and the charcoal was added to the resulting solution. The mixture was stirred for 30 min at 85 °C, and filtered through Celite® while still hot. The desired compound was collected by removing the solvent from the filtrate. This procedure was used to purify the intermediates as well as the products from monomer syntheses reported below.Synthesis of 5,6-dihydroxy-1H-benzimidazole
The procedure was modified from that of Weinberger et al.46 Formic acid (99.9%, ca. 30 mL) was added dropwise to 1,2-diamino-4,5-dimethoxy-benzene (1.0 g, 5.95 mmol) and the resulting mixture was refluxed for 2 h at 100 °C. Hydrobromic acid (48%, ca. 30 mL) was then added to the reaction mixture and refluxed again for 8 h at 100 °C. Upon completion, the reaction mixture was chilled over an ice-water bath. The precipitate formed was collected and purified with decolorizing charcoal as described to obtain the beige product (58%): δH (300 MHz; DMSO-d6), 7.12 (2H, s, Ar H), 9.25 (1H, s, CH) ppm. δC (75 MHz; DMSO-d6), 98.37 (C-4, C-7), 123.6 (C-8, C-9), 136.9 (C-2), 146.4 (C-5, C-6) ppm.Synthesis of 2-methyl-5,6-dihydroxy-1H-benzimidazole
The procedure was developed from the work of El-Nezhawy et al. and Weinberger et al.46,47 Glacial acetic acid (2.00 mL, 35.0 mmol) was added dropwise to 1,2-diamino-4,5-dimethoxy-benzene (1.0 g, 5.9 mmol) dissolved in hydrochloric acid (4 N, 8.0 mL) and the resulting mixture was refluxed for 4 h at 100 °C. The reaction mixture was then chilled over an ice-water bath and further concentrated by reducing the volume with an air jet. The precipitate formed was filtered out and purified with decolorizing charcoal to obtain the intermediate. Excess hydrobromic acid (48%, ca. 35 mL) was added to the intermediate and the mixture was refluxed for 8 h at 100 °C. Upon chilling, a precipitate formed in the reaction mixture. The precipitate was purified with decolorizing charcoal and the pale purple product was collected after solvent removal (39% yields): δH (300 MHz; DMSO-d6), 7.02 (2H, s, Ar H), 2.68 (3H, s, CH3) ppm. δC (75 MHz; DMSO-d6), 12.08 (C-10), 98.24 (C-4, C-7), 123.8 (C-8, C-9), 145.6 (C-5, C-6), 147.5 (C-2) ppm.Synthesis of 2-trifluoromethyl-5,6-dihydroxy-1H-benzimidazole
The procedure was developed from the work of René et al. and Weinberger et al.46,48 A 0.5 mM reaction mixture of 1,2-diamino-4,5-dimethoxybenzene (1.0 g, 5.9 mmol) in trifluoroacetic acid (12 mL) was prepared over an ice-water bath, which was then refluxed at 70 °C for 16 h. Upon completion, the acid was removed under reduced pressure. The residue was purified with decolorizing charcoal. Excess hydrobromic acid (48%, ca. 35 mL) was added to the intermediate obtained and the mixture was refluxed for 8 h at 100 °C. Upon chilling, an off-white/grey precipitate formed in the reaction mixture, which was collected as the product (25% yields): δH (300 MHz; DMSO-d6), 6.99 (2H, s, Ar H) ppm. δC (75 MHz; DMSO-d6), 100.3 (C-4, C-7), 131.0 (C-8, C-9), 142.4 (C-10), 145.3 (C-5, C-6), 145.9 (C-2) ppm.Self-polymerization of the synthesized monomers and preparation of PDHBI-coated substrates
DHBI (300.28 mg, 2.00 mmol) was dissolved in a buffer solution of pH 10 prepared from TRIS (242.4 mg, 2.00 mmol) dissolved in deionized water (200 mL).34 The resulting solution would have a final pH of 8.5, which was stirred for 24 h at ambient conditions. The grey-green polymer precipitated out from solution and was collected by Millipore filtration. It was then rinsed with small portions of water to remove residual TRIS. Similar procedure was performed with the other two monomers based on the same molar content. It is notable that the polymer of 2-CF3-DHBI [P(2-CF3-DHBI)] is water soluble. Thus the TRIS buffer and unreacted material were removed by dialysis against deionized water, and the polymer was obtained by freeze-drying the dialyzed solution.A similar procedure was adopted to prepare PDHBI-coated silica particles (PDHBI@SiO2) in situ during the self-polymerization by dispersing the particle templates (500 mg) in the reaction mixture.34 After 24 h, the surface-coated particles were separated from non-surface-bound materials by centrifugation (3000 rpm, 5 min) and washing with deionized water for several cycles until the supernatant became clear and colorless. The particles were then filtered and rinsed with deionized water. An equimolar mixture of dopamine and DHBI (i.e. 1 mmol each) was used for the preparation of copolymer-coated silica particles [P(DHBI-DA)@SiO2]. PDHBI was coated on PET film, stainless steel plate and Millipore filter sheet in a similar manner.
Kinetics study of the self-polymerization of 5,6-dihydroxy-1H-benzimidazole by dynamic light scattering
The procedure was the same as for preparing PDHBI@SiO2. The reaction mixture was divided into eight fractions of equal volume prior to the 28 h reaction period. The fractions were taken off accordingly after 1, 3, 6, 10, 14.5, 17.5, 24 and 28 h, and purified accordingly to retrieve the kinetics samples. The same procedure was performed with dopamine to prepare kinetics samples of PDA-coated silica particles (PDA@SiO2) for comparison. Measurements were performed to determine the change in particle size over time.Ultraviolet-stimulated self-polymerization of 5,6-dihydroxy-1H-benzimidazole
A reaction mixture of DHBI and TRIS was prepared as described previously. The volume was divided equally into two round bottom flasks. Both flasks were exposed to UV-B source (Sankyo-Denki G8T5E) irradiated from the CL-1000 ultraviolet crosslinker (UVP), while one of the flasks was covered with aluminium foil and acted as a control. A small aliquot (16 µL) of the reaction mixture was sampled each time and diluted by 3 mL of deionized water for absorbance measurement with an UV-Vis spectrophotometer. Samples were taken every hour for 7 h, while the last sample was taken after 24 h.Characterization
1H, 13C, 19F and 2D nuclear magnetic resonance (NMR) spectroscopies were performed on a Bruker Avance III 300 MHz spectrometer. 13C solid-state (CP-MAS) spectroscopy was performed on a Bruker Avance III 300 Solid State spectrometer. Cyclic voltammetry (CV) analyses were performed with an eDAQ Integrated Potentiostat System (ER466) equipped with an Ag/AgCl reference electrode, and platinum counter and working electrodes. Dynamic light scattering (DLS) and zeta potential measurements were performed on a Malvern Zetasizer Nano. Thermogravimetric analysis (TGA) was performed on a Q5000 (V3.15 Build 263) instrument purchased from TA Instruments. Transmission electron microscopy (TEM) images were acquired with a JEOL 1400 transmission electron microscope. Electron Paramagnetic Resonance (EPR) spectroscopy was performed on a Bruker EMX-Plus X-Band ESR Spectrometer. Gel permeation chromatography (GPC) was performed by a unit purchased from Shimadzu Scientific Instruments, and the chromatography columns were purchased from Phenomenex. Contact angle measurements were performed using a CAM200 Contact Angle and Surface Tension Meter. Ultraviolet-visible (UV-Vis) spectrophotometry was performed on a Varian Cary 300 UV-Visible Spectrophotometer. Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy was performed on a Bruker IFS 66/S single-beam spectrometer.Results and discussion
A three-step procedure has been employed to synthesize 5,6-dihydroxy-1H-benzimidazole as the initial monomer for testing (Scheme 3). It formed the basis of the present work for its simple structure, when compared to the other DHBI-based monomers prepared, and the highest resemblance to DHIn. 2D NMR was performed on DHBI to identify the potential reactive sites during the self-polymerization process (Fig. S1†). The monomer is soluble in TRIS buffer and compatible with dopamine as a comonomer in the reaction mixture. Hence, we were able to prepare materials based on PDHBI and the DHBI-dopamine copolymer [P(DHBI-DA)] in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio for further study.
:1 molar ratio for further study.
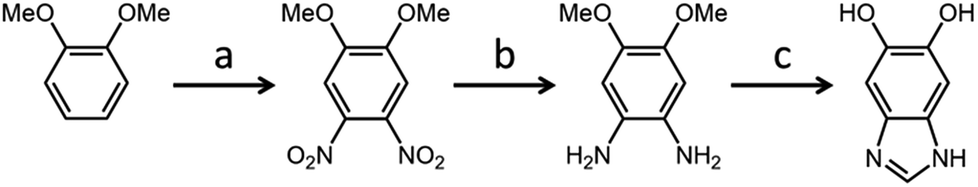 | ||
| Scheme 3 The synthesis of 5,6-dihydroxy-1H-benzimidazole: (a) (i) – nitric acid (65%), 0 °C; (ii) – 80 °C, 2 h, (b) Pd/C, hydrazine monohydrate, methanol, reflux at 50 °C, 1 h, (c) (i) – formic acid (99.9%), reflux at 100 °C, 2 h; (ii) – hydrobromic acid (48%), reflux at 100 °C, 8 h. | ||
We initially suspected that DHBI-based materials would be cross-linked like PDA since the hydrogen at the C-2 carbon of DHBI is potentially labile, which is similar to that of DHIn,19,20 and not possible to be analyzed by solution-state analysis. However, to our surprise, we were able to dissolve a small fraction of PDHBI in dimethyl sulfoxide (DMSO), which immediately suggested that the PDHBI possesses a lower degree of cross-linking than PDA. The difference in cross-linking density between the two polymers can be justified through comparing the respective monomers with regard to molecular structure and reactivity of the five-member ring. Firstly, it has been reported in the literature that C-3 of DHIn also plays a minor role in forming linkages with another monomer unit during the polymerization although C-2 is the preferential active site for cross-linking.21,53,54 In contrast, DHBI lacks such structural advantage since its C-3 has been occupied by the sp2 hybridized nitrogen, which in turn leads to fewer linkage formations. In terms of reactivity, both the pyrrole and imidazole moieties behave as nucleophiles as the lone electron pair of the sp3 hybridized nitrogen resonates among all other positions on the ring to induce an overall negative charge.55 They would attack the catechol moiety, which behaves as an electrophile during the polymerization, of another monomer unit at C-4 and C-7.56 Such reaction regime is particularly probable for DHIn since the asymmetrical structure and different acidity of its hydroxyl groups direct electron density towards C-2, making it a stronger nucleophile,21 whereas the symmetrical structure of DHBI might not be able to achieve the same effect on the reactivity at the corresponding C-2. The overall effect of the differences discussed above contributes to the lower cross-linking density of PDHBI compared to PDA.
In order to understand the reaction pathway taken by DHBI during the self-polymerization and elucidate the similarities and differences to that of dopamine, PDHBI was partially dissolved in deuterated DMSO and analyzed by 1H NMR. The soluble fraction was collected through a syringe filter and contained just enough material to acquire appreciable signal strength. Substantial magnification was needed to illustrate the significant peaks of the PDHBI spectrum since the signals originated from the residual 1H at the unreacted positions, if the polymer is structurally similar to PDA. By comparing the 1H spectrum of PDHBI against that of the monomer (Fig. 1a), the peak (9.25 ppm) of the 1H at C-2 of DHBI (Ha) is not observed in the PDHBI spectrum, but a newcluster of peaks has emerged at 8.00–8.50 ppm. The cluster was attributed to residual unreacted hydrogens at the C-2 carbon incorporated into the polymer, subject to a different chemical environment. Furthermore, the peak (7.12 ppm) of the 1H at C-4 and C-7 of DHBI (Hb) has exhibited some degree of broadening and reduction in the integral ratio with the other significant peaks – the ratio between Hb and Ha is 2:1 for DHBI, whereas that between Hb and the cluster is 2:7 for PDHBI. The lower number of Hb in the polymer suggests that C-4 and C-7 are reactive during the self-polymerization. It is notable that the chemical shift for Hb has not changed significantly, which is likely due to these residual hydrogens only existing on the polymer chain ends.
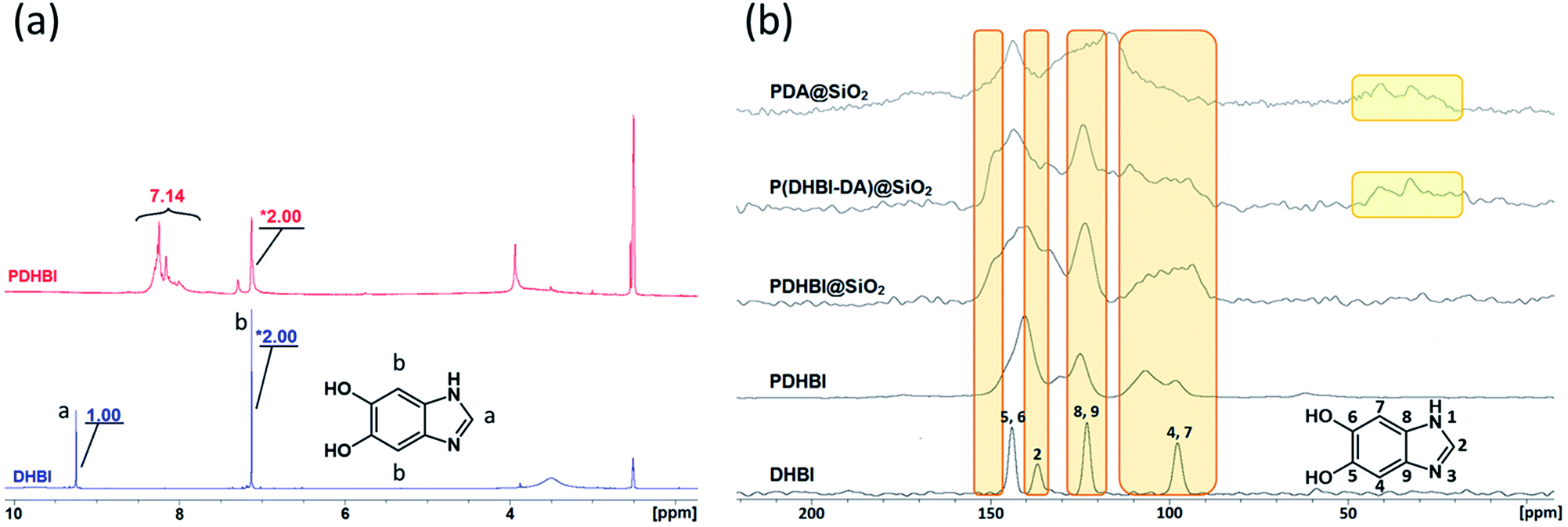 | ||
| Fig. 1 Comparison of NMR results acquired for the synthesized materials: 1H spectra of DHBI and PDHBI (a) and 13C solid-state NMR spectra of DHBI, PDHBI, PDHBI@SiO2, P(DHBI-DA)@SiO2 and PDA@SiO2 (b). | ||
The results above are in agreement with the accepted model for the self-polymerization of dopamine, and other heterocyclic catechol derivatives in general.19,20 However, the 1H NMR spectra was not definitive proof for the lightly cross-linked nature of PDHBI, i.e. to prove that C-2 of DHBI is a reactive site. Hence, 13C solid state NMR (CP-MAS) was performed to elucidate the reactivity of the C-2, C-4 and C-7 carbon in order to gain further understanding of the polymerization mechanism for DHBI and the structure of PDHBI. Fig. 1b compares the 13C spectra between the DHBI monomer and its homopolymer, as well as PDHBI@SiO2 and P(DHBI-DA)@SiO2. All of the polymeric samples (including PDA@SiO2 for comparison) exhibit extensive peak broadening, indicating an extended polymeric architecture. The PDHBI homopolymer peaks are sharper than the other samples polymerized on the silica template due to the increased polymer concentration and subsequent signal-to-noise reduction. The DHBI monomer exhibits a sharp peak for the C-4 and C-7 carbons at 97 ppm. When polymerized, significant peak broadening and intensity reduction of the C-4 and C-7 peaks for PDHBI and PDHBI@SiO2, located between 90 and 110 ppm, is observed. This confirms that reactions have taken place at these carbons, consistent with carbon–carbon bond formation, generating a π-conjugated system with a large number of repeating units, as shown in Scheme 1. Such a structure is analogous to PDA materials previously reported.19,20 Peak broadening at 90–110 ppm is also exhibited by P(DHBI-DA)@SiO2, whereas when dopamine is polymerized alone on silica, such peaks are not evident. This result indicates that the copolymer P(DHBI-DA) successfully incorporates PDHBI into the PDA structure.
Results gathered from 13C solid state NMR confirms that the C-2 carbon on DHBI, represented by the small peak at 137 ppm, is indeed reactive enough to facilitate cross-linking reactions during the polymerization. As indicated by the 13C spectra of the DHBI-containing polymer samples (Fig. 1b), the C-2 peak has both broadened and shifted upfield. The reactivity of the C-2 carbon does not only suggest that the polymerization of DHBI is able to yield materials analogous to PDA (Scheme 1),19 it also explains the poor solubility of PDHBI mentioned in previous discussion.
Besides the reactive carbons, the non-reactive quaternary carbons (C-8 and C-9) are represented by a sharp peak which arises at 123 ppm. The peak is present in DHBI and all DHBI-containing polymer samples, i.e. PDHBI, PDHBI@SiO2, and P(DHBI-DA)@SiO2, while its intensity and peak shape remain relatively unchanged. On the other hand, the catecholic carbons (C-5 and C-6) exhibit peak broadening in all PDHBI polymeric samples, centering around 145 ppm. The broadening is more pronounced for PDHBI@SiO2 and P(DHBI-DA)@SiO2, which could arise from the interaction of the catechol groups with the silica surface. The peak broadening exhibited by C-5 and C-6 carbons is not as pronounced as that displayed by C-4 and C-7, due to their non-linking nature in the overall polymer. It is interesting to note that no appreciable peaks are present downfield of 160 ppm for the PDHBI samples, indicating that there are low levels of oxidized carbonyl groups present in the polymer. This is in contrast with PDA, which exhibits a cluster of weak and broad peaks from 160–180 ppm attributed to carbonyl groups (Fig. S2†).
The NMR results indicate a high degree of structural similarity shared between PDHBI and PDA, which suggests that self-polymerization of DHBI proceeds in the same manner as dopamine, facilitated by the redox chemistry when dissolved in an aqueous buffer at ambient conditions (Scheme 1). To prove our hypothesis, the electrochemical properties of the DHBI and dopamine polymerization were compared using CV. Control experiments with only TRIS buffer solution without the monomer yielded no electrochemical activity. The polymerization of DHBI showed strong evidence of oxidation with the reduction potential peaking at approximately 0.46 V, associated with the formation of a grey-green precipitate. Similar results were obtained for the dopamine polymerization, with the reduction potential peaking at approximately 0.48 V, accompanied by the formation of a fine, black precipitate. Considering the electrochemical similarity of the two reactions, we would expect that DHBI and dopamine share the same redox-facilitated self-polymerization mechanism. As shown by the voltammogram (Fig. 2a), both DHBI and dopamine polymerizations are irreversible since no reduction peak was observed as the current was reversed. While differences in the diffusion coefficients were believed to be the cause of the different cathodic currents required to initiate the oxidation of DHBI and dopamine, more in-depth study would help to elucidate how the current influences the redox reactions. The working electrode was quickly coated with a thin layer of polymeric material after the first few CV cycles which impeded further measurements. The polymer coating formed more rapidly with DHBI than with dopamine, which suggests faster reaction kinetics for DHBI. To gain further insight, kinetics study was performed by using dynamic light scattering (DLS) to determine the thickness growth of the respective polymer coating on fumed-silica particle templates over a 28 hour period (Fig. 2b). The growth of PDHBI was found to be significantly faster, which plateaued at 50 nm within 10 h while it took 24–28 h for PDA to reach the same thickness.‡ A similar trend was observed from the degradable (organic) content of the kinetics samples when investigated by TGA (Fig. 2c). Notably, PDHBI-coated samples exhibited a significantly higher degradable content than PDA-coated samples with the same coating thickness/polymerization time, meaning that PDHBI would be a denser material than PDA. The result is also reflected in the TEM images, in which a dense, uniform coating of PDHBI partially blocked the electron beam from reaching the silica particle templates (Fig. 3).
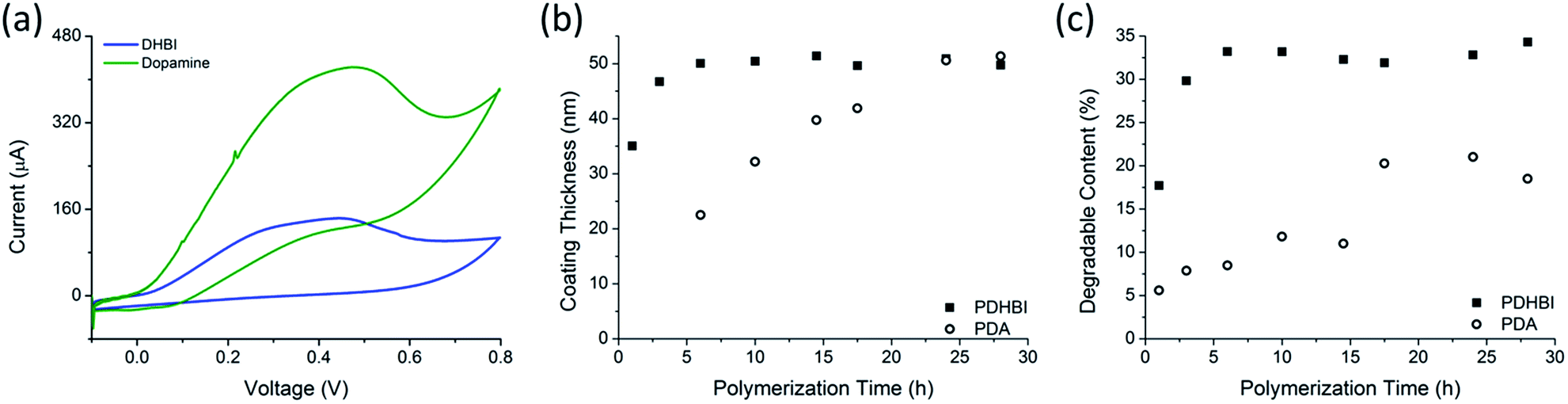 | ||
| Fig. 2 Study of the self-polymerization of DHBI and dopamine by cyclic voltammetry (a) and elucidation of the individual polymerization kinetics by monitoring the growth of the polymer coating on silica particles with respect to changes in thickness (b) and amount of degradable organic content (c) over the course of the polymerization. | ||
 | ||
| Fig. 3 TEM images of silica particle templates coated with PDHBI [scale bar = 100 nm (top) and 0.2 µm (bottom)]. Note that the small particles in the background were residual nano-size silica. | ||
Although DHBI is able to self-polymerize in a faster manner than dopamine, it is possible to shorten the reaction time further. Sheng et al. have recently demonstrated the stimulation of the self-polymerization of dopamine by UV irradiation.50 Since DHBI exhibits the same polymerization mechanism as dopamine, there is a potential to apply UV-stimulation to this synthetic monomer. As a result, self-polymerization of DHBI was conduct under UV irradiation and monitored over a 7 h period. Progressive changes in color and turbidity/translucency have been observed from both the control (Fig. S3a†) and the UV-stimulated (Fig. S3b†) samples. The turbidity of the UV-stimulated sample increased significantly faster and the reaction mixture lost its translucency within 6 h, whereas the control was still relatively clear after 7 h, indicating an enhanced reaction rate by UV irradiation. Moreover, the UV irradiated sample displayed a darker green color (Fig. S3c,† right) compared to the control (Fig. S3c,† left) after 24 h. The darker color of the UV-stimulated sample suggested a higher amount of PDHBI was generated. Since PDA and its analogues absorb strongly in the visible light and UV region of the electromagnetic spectrum,34,50 UV-Vis spectrophotometry was performed as a means of monitoring changes during the UV-stimulated polymerization with comparison made against the control sample. The spectra obtained for the control (Fig. 4a) and UV-stimulated (Fig. 4b) samples showed absorption peaks appearing at 249 and 294 nm – within the region (200–300 nm) that represents the generation of PDA analogue.57 The change in absorbance at 294 nm, in particular, has been plotted to further elucidate the influence of UV irradiation on the self-polymerization kinetics of DHBI (Fig. 4c). The plot showed that the UV-stimulated sample increased faster in absorbance than the control by approximately an hour, which then began to decrease at 7 h, while that for the control was still at its maximum. The decrease in absorbance was expected as PDHBI particles/aggregates settledmore rapidly as they grew larger, and less material should be detected as a result. Note that the absorbance of UV-stimulated sample remained relatively unchanged at 7 and 24 h in comparison to the control, indicating that the time required for the self-polymerization of DHBI to approach completion can be shorten under UV stimulation. Therefore, UV irradiation can be used as stimulation for the preparation of PDHBI. However, the irradiation has no influence on the chemical nature on the degree of cross-linking of the resulting polymer as indicated by the highly similar ATR-FTIR spectra and thermal degradation traces for the irradiated and non-irradiated samples (Fig. S4†).
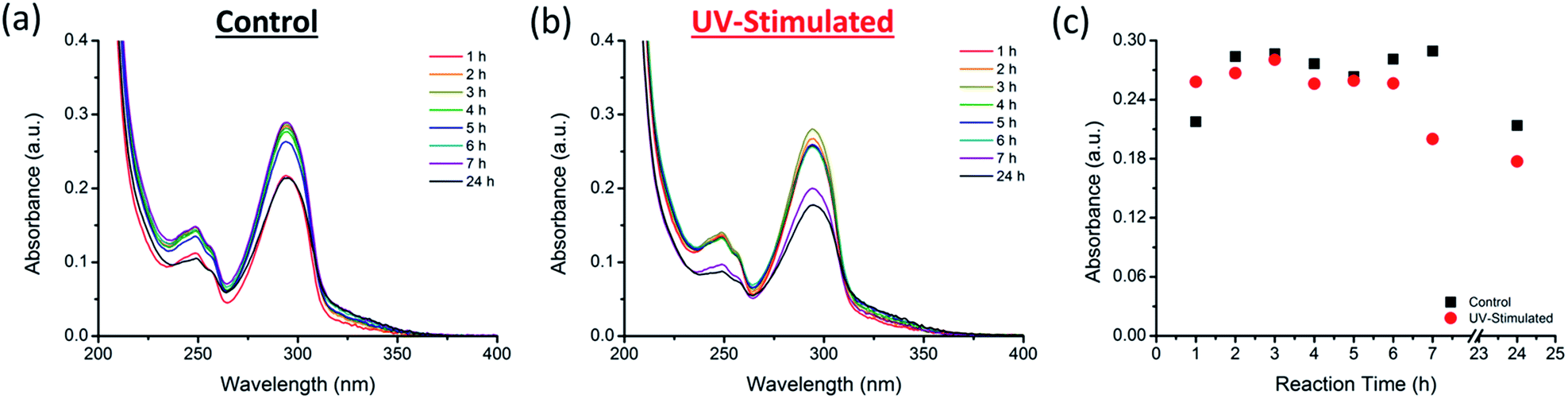 | ||
| Fig. 4 UV-Vis spectra acquired for the control (a) and the UV-stimulated (b) samples during the course of the self-polymerization of DHBI, and the changes in absorbance at 294 nm plotted against time (c). | ||
Beside the similar polymerization mechanism, DHBI-based polymeric materials also possess several intrinsic properties which are found in PDA. It has been reported that PDA carries organic free radicals within the structure like its biological analogues melanin and eumelanin.58–60 Under specific conditions, PDA-coated substrates can be used to initiate radical polymerization.50 Inspired by such interesting phenomenon, EPR analysis was performed on both PDHBI and P(DHBI-DA) supported on silica particles. It was found that both samples contained a stable organic free-radical in a similar fashion to PDA. A single peak at 3508 G (g-factor = 2.00335) was generated due to the presence of randomly orientated π-electron free radical species, which was consistent with results obtained for PDA (Fig. 5a), both in this study and in the literature.58 The EPR signal for these materials is suspected to arise from at least two different types of radicals: defects present in the polymer backbone, and a second type likely due to an o-benzosemiquinone anion radical.59 Quinone formation in PDHBI has been proven by ATR-FTIR, in which a weak, broad absorption peak was observed in the 1800–1600 cm−1 region of the spectrum (Fig. 5b), though the quinone structure is less prevalent than in PDA, as shown by 13C solid state NMR. The EPR results helped further elucidate the structure of the prepared polymers, of which the presence of free radicals reflects an irregular cross-linked polymer network with a variety of bonding arrangements.9,61
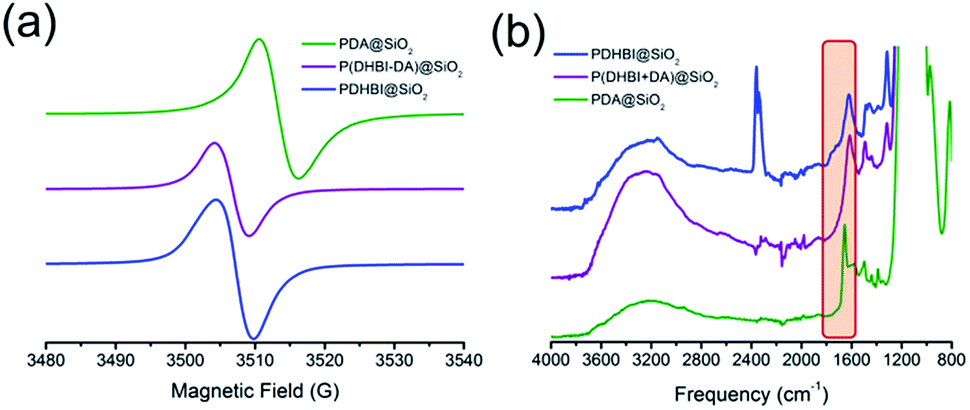 | ||
| Fig. 5 EPR (a) and ATR-FTIR (b) spectra of silica particle templates coated with PDHBI, P(DHBI-DA), and PDA. | ||
PDHBI is also similar to PDA in terms of surface modifying capability. Coating of PDHBI can be formed in situ for direct surface modification with not only silica particles but also other substrates present in the reaction mixture. Coating has been performed on Millipore filter sheet, stainless steel plate, and PET films. All coated materials exhibited visible film coating (Fig. S5†), as well as ATR-FTIR bands similar to those observed for the coated silica particles (Fig. S6†). The change in surface wettability of the coated materials was assessed using static contact angle measurements (Table S1†). A marked reduction in contact angle was observed for every coated sample compared with the naked substrate. While the coated stainless steel plate and Millipore filter sheet exhibited contact angles of less than 50°, the PET film was only reduced to 73°, attributed to the uneven nature of the PET film. In each case, the ability to coat various substrates and increase the hydrophilicity of the substrates is in line with that observed for substrates coated with polymers of dopamine and other catecholamines.1,26,62
Although PDHBI is capable of coating on various surfaces like PDA, the resulting coating is expected to be less thermally stable than that of PDA due to the lower degree of cross-linking of the DHBI-based polymer as discussed previously. In order to gain further insight into the thermal degradation differences of the coatings without substrate influences, free PDHBI and PDA were prepared for TGA studies. As illustrated by Fig. S7a,† PDHBI indeed degraded at a lower on-set temperature (287 °C) than PDA (322 °C). When the homopolymers were applied as a coating on silica particles, a similar trend was observed from the degradation (Fig. S7b†). We further investigated the possibility to enhance the thermal properties of PDHBI-related materials by introducing dopamine as a comonomer to promote cross-linking, of which we have demonstrated with P(DHBI-DA)@SiO2. The incorporation of dopamine increased the cross-link density for the resulting polymer, and thus enhanced the thermal stability as indicated by the comparable on-set degradation temperature to PDA coating and slower degradation below 450 °C in contrast to PDHBI. It is notable that PDHBI, as both free polymer and coating, exhibited better resistance to high temperatures than PDA as it degraded more slowly from 500 °C onward. The copolymer, on the other hand, exhibited only slightly better resistance at high temperatures compared to PDA, but not to the same extent as PDHBI. It was believed that the thermal resistance of PDHBI towards high temperatures is attributed to its monomer unit, i.e. the benzimidazole origin, which is more abundant in the homopolymer than in the copolymer.
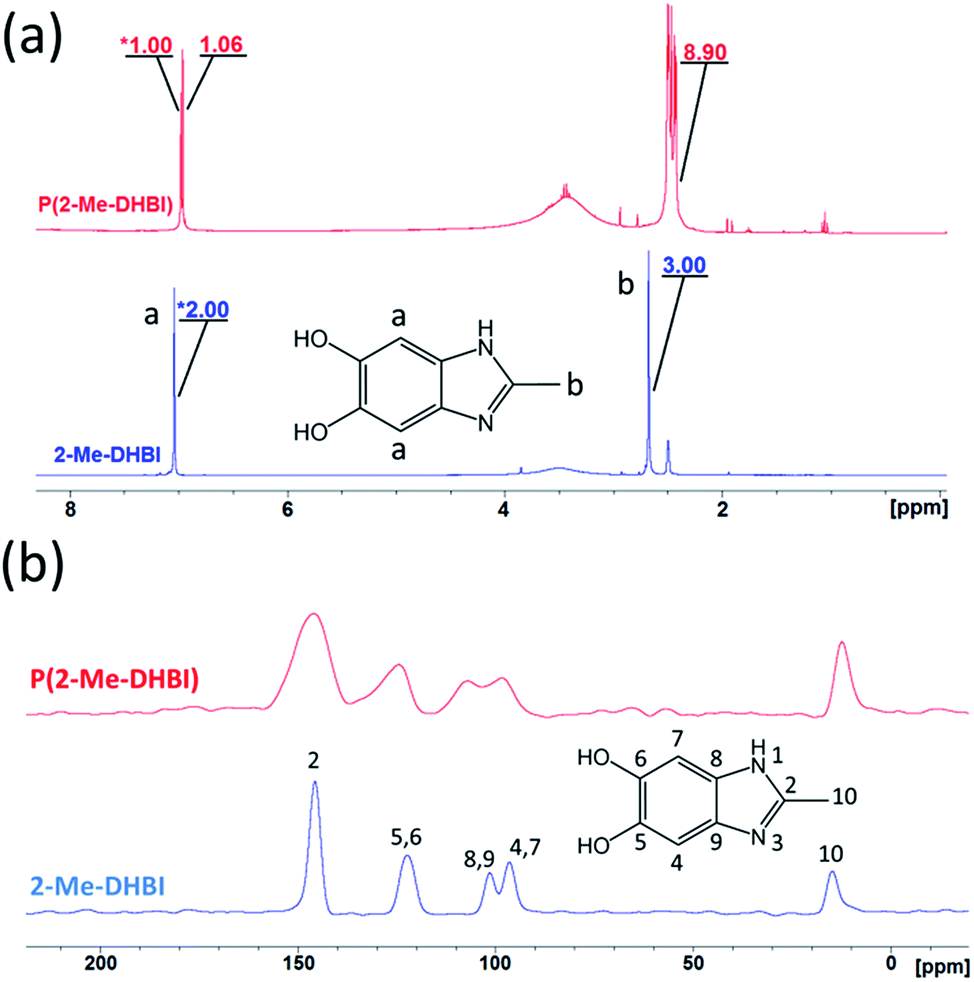
Copolymerization is readily achievable, allowing structural modification to manipulate certain properties of PDHBI. However, the imidazole ring can be easily modified during the monomer synthesis process to yield a range of DHBI-based species with tailored functionalities at the 2-carbon position, which can also result in PDHBI analogues of manipulated properties. As a proof of concept, we utilized the reaction between o-benzenediamine and a range of carboxylic acids as a versatile platform to prepare 2-substituted DHBI monomer (Scheme 2).49,63–65 There are still technical barriers to be overcome with di- and tricarboxylic acids, as well as aromatic acids in terms of reaction conditions and purifications. With simple aliphatic acids such as acetic acid and trifluoroacetic acid, in contrast, we successfully synthesized 2-Me-DHBI and 2-CF3-DHBI, respectively. 2D NMR has been performed to elucidate the respective molecular structure (Fig. S8 and S9†). Fig. 6a illustrated a comparison of the ATR-FTIR spectra between all the DHBI-based monomers, in which the regions represent the common and unique molecular structures have been highlighted. These monomers were able to self-polymerize under the same reaction conditions for the preparation of PDHBI. The broadening effect of the absorption peaks because of the increased level of interactions after the self-polymerization was observed from all the polymer spectra,66 indicating the successful preparation of P(2-Me-DHBI) and P(2-CF3-DHBI) (Fig. 6b).
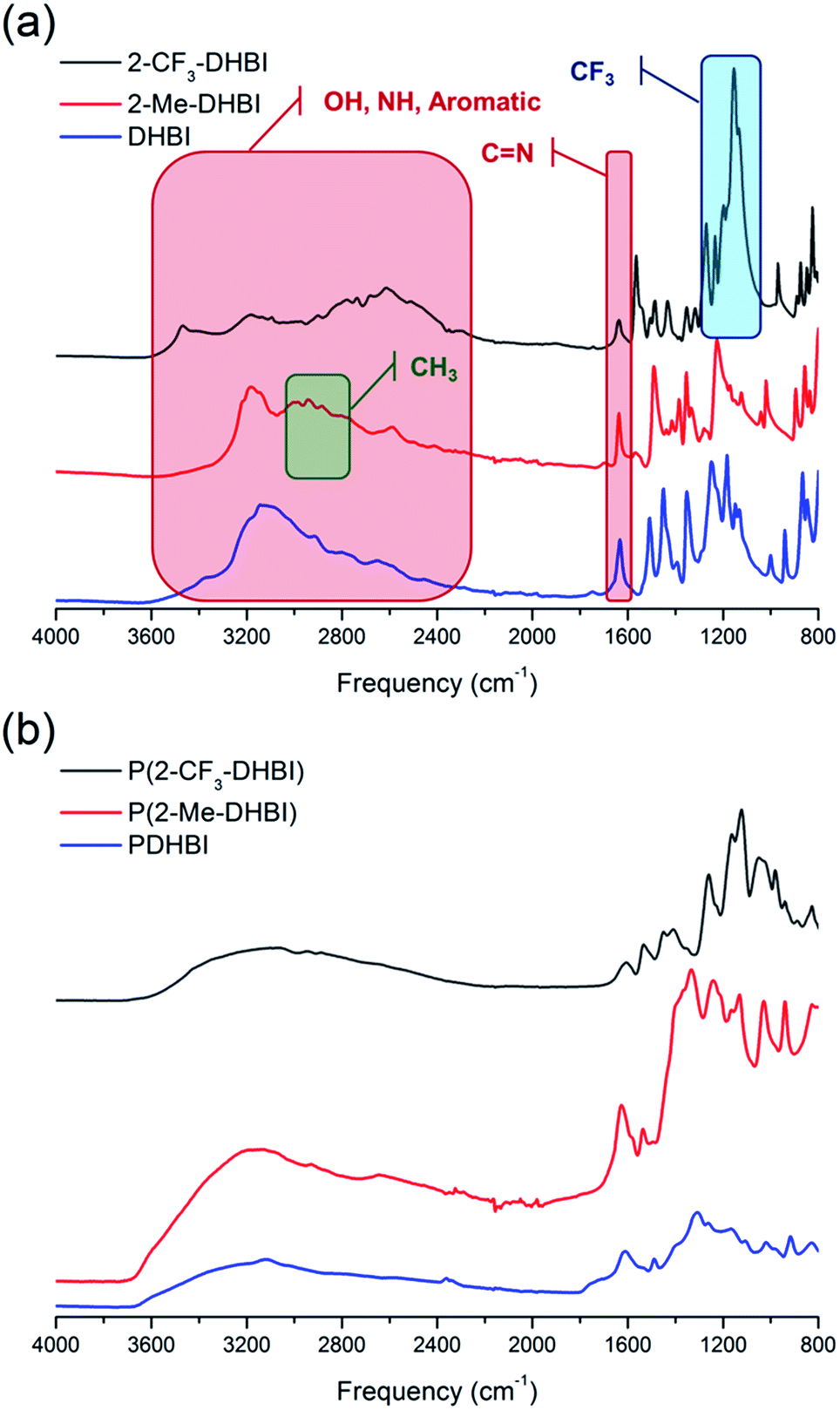 | ||
| Fig. 6 ATR-FTIR spectra of DHBI, 2-Me-DHBI, 2-CF3-DHBI (a) and the corresponding polymers (b). | ||
Despite the common DHBI origin, immediate differences were observed in the self-polymerization of the 2-CF3-substituted monomer, namely that a color change was observed whilst no precipitate was formed. The reaction mixture of 2-Me-DHBI, in contrast, also exhibited a color change but a dark green precipitate was formed, which was similar to the visual changes observed when DHBI self-polymerized. It can be confidently stated that P(2-Me-DHBI) and P(2-CF3-DHBI) would not be able to cross-link like PDHBI because the 2-carbon position has been occupied and averts the formation of linkages, i.e. the 2-substituted polymers are linear in structure. Despite the structural similarity, the key to the difference in solubility between P(2-Me-DHBI) and P(2-CF3-DHBI) was believed to be the C-2-substituent. The CF3 group would be able to induce a significantly stronger polarization effect on the overall structure than the methyl group because of the highly electronegative fluorine atoms, which also helps to establish hydrogen bonding with the surrounding water molecules. As a result, the more polar P(2-CF3-DHBI) stayed soluble in the TRIS buffer after self-polymerization while P(2-Me-DHBI) precipitated out. This preliminary observation effectively demonstrated that the properties of PDHBI can be manipulated by tailoring the functionality at the 2-carbon position.
With the 2-substituted polymers, we have performed NMR analyses in a similar manner as with PDHBI. Although P(2-Me-DHBI) is solely linear and more soluble than PDHBI, some material could not dissolve into DMSO and was believed to be due to its higher molecular weight. P(2-CF3-DHBI), in comparison, was completely soluble in DMSO because of its higher solubility than the other two polymers in the chosen solvent. Similar to PDHBI, P(2-Me-DHBI) exhibit changes in its 1H spectrum compared to the monomer (Fig. 7a). Substantial magnification was also needed for the signals because of the low concentration of P(2-Me-DHBI) in the sample even though a significant quantity of the polymer was added. Although being non-reactive in nature, the methyl group at C-2 represented by the singlet at 2.68 ppm was shifted up field to about 2.45 ppm and split into a cluster of peaks which overlapped with the solvent peak. The split is attributed to the increased interactions with neighboring 1H because of the structural confinement resulted from the polymerization. The peak of the 1H at C-4 and C-7 of 2-Me-DHBI (7.02 ppm) was also split into two singlets of the same integral and shifted up field slightly. The significant decrease in the integral ratio between this peak and the methyl peak (from 2:3 to almost 2:9) after the polymerization suggests the displacement of 1H from C-4 and C-7. Hence, the acquired signals solely originated from the residual 1H present at the chain ends of P(2-Me-DHBI). 13C NMR spectrum was also obtained, but it required the solid-state technique due to the insufficient signal strength available from the dissolve polymer. Fig. 7b illustrates the typical peak broadening/shifting phenomenon to C-4 and C-7 after the polymerization. The C-8 and C-9 peak was also slightly distorted because of the merging of the C-4 and C-7 peak. The peaks of C-2, C-5 and C-6, and C-10 only broadened in contrast. The unchanged chemical shift of C-2 further indicates its non-reactive nature, which in turn suggests that the occupied 2-carbon position would promote linear chain growth for P(2-Me-DHBI) through π-conjugations.
 | ||
| Fig. 7 Comparison of the 1H NMR (a) and 13C solid-state NMR (b) spectra of 2-Me-DHBI and P(2-Me-DHBI). | ||
In comparison, the higher solubility of P(2-CF3-DHBI) in DMSO allowed better 1H and 13C spectra to be obtained with solution NMR. Splitting of the 1H peak of C-4 and C-7 (6.99 ppm) was also observed after the self-polymerization of 2CF3-DHBI (Fig. S10†). The resulting cluster gave evidence of π-conjugations occurred between monomer units, which subjected the residual 1H on C-4 or C-7, i.e. the chain ends, to different chemical environments. For the 13C spectrum of P(2-CF3-DHBI) (Fig. S11†), clusters of peaks have emerged, suggesting an increased level of interactions which typically happens after the self-polymerization.
We were able to associate the residual 1H to their host and neighboring carbons readily by 2D NMR credited to the similar pattern of proton-to-carbon interactions exhibited by P(2-CF3-DHBI) and its monomer (Fig. 8). Attributed to the better resolution of 13C solution NMR than that of the solid state technique, the results allowed us to confirm the presence of at least three types of chemically different chain ends in the polymer sample. The 13C peaks that did not show any interaction with the residual 1H, except those of C-10, were believed to arise from the carbons of the monomer units at the center of P(2-CF3-DHBI). With C-10, 19F NMR analysis was performed to verify changes occurred to the trifluoromethyl group after the self-polymerization of 2-CF3-DHBI.
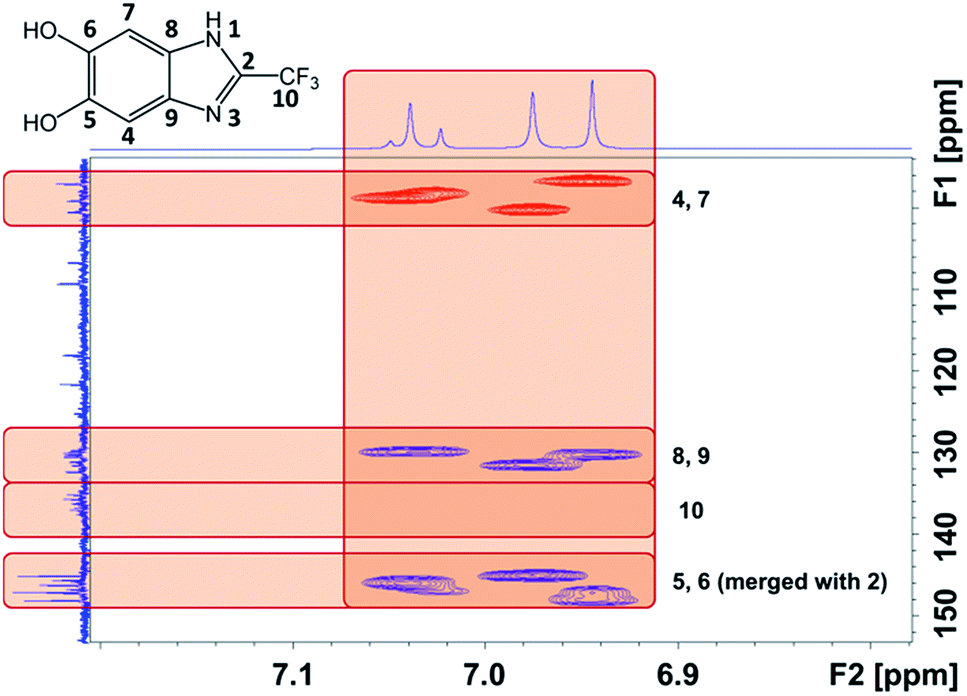 | ||
| Fig. 8 2D NMR spectrum of P(2-CF3-DHBI) acquired by heteronuclear single-quantum correlation (HSQC, red signals) and heteronuclear multiple-bond correlation (HMBC, blue signals) techniques (horizontal = 1H, vertical = 13C). | ||
As illustrated by Fig. S12,† the singlet of the monomer was split into a cluster of three peaks, i.e. three new chemical environments, which is in agreement with the aforementioned results. It should be noted that there is a strong aliphatic signal at 3.44 ppm on the 1H spectrum of P(2-CF3-DHBI), which has a corresponding signal at 60.2 ppm on the 13C spectrum (Fig. S10 and S11†). The origin of these aliphatic signals was believed to be the residual methoxy groups on some monomer units resulting from incomplete demethylation. Since the methoxy 1H would not be displaced during the self-polymerization, the signal became prominent relative to the residual 1H on C-4 and C-7 when incorporated into the polymer. Nonetheless, the presence of residual methoxy groups did not prevent 2-CF3-DHBI from self-polymerizing.
In brief, the NMR results of P(2-Me-DHBI) and P(2-CF3-DHBI), again, indicated the successful preparation of these polymeric materials. These results also strengthened the argument that the π-conjugation at C-4 and C-7 is the dominant mechanism to facilitate the self-polymerization of heterocyclic catechol derivatives, which in turn gives flexibility on the choice of C-2-substituent for tailoring the performance of the resulting polymer.
The increased solubility of the newly prepared materials allowed us to further unravel the less studied polymeric nature of these analogues of PDA, which would not be possible with catecholamine-based materials due to their poor solubility in most solvents. Samples were prepared for GPC in N,N-dimethylacetamide (DMAc) with the soluble fraction of PDHBI and P(2-Me-DHBI), and P(2-CF3-DHBI). The molecular weight (MW) distribution of the respective polymer was illustrated in Fig. 9a. The MW (Mn and Mw) and dispersity (Đ) of the samples have been reported on Table S2.† The uncontrolled nature of the redox-facilitated self-polymerization technique resulted in a broad distribution for all polymers, of which P(2-Me-DHBI) exhibited the widest range of MW (Đ = 2.43), followed by PDHBI and P(2-CF3-DHBI) (Đ = 1.46 and 1.30, respectively). As discussed previously, PDHBI has a lightly cross-linked structure. The effect of cross-linking increases with MW, which reduces the solubility of the high-MW fraction. As a result, only the least cross-linked, low-MW fraction (Mw ∼ 23000 g mol−1) was present in the GPC sample. In contrast, P(2-Me-DHBI) is linear and its solubility is more likely to be restricted by MW, i.e. the chain length, in spite of its methyl functionality. Hence, fraction of higher MW (Mw ∼ 37000 g mol−1) was able to dissolve into DMAc. It was expected that P(2-CF3-DHBI) would exhibit a MW distribution similar to P(2-Me-DHBI) because of their almost identical monomer units, but the 2-CF3-substituted polymer has a lower MW and also a narrower dispersity. This notable contrast could possibly be attributed to the difference in electron density induced by the respective C-2-substituent. For instance, the electronegative fluorine atoms would have withdrawn electrons from the aromatic ring and limited its reactivity. It is also possible that the CF3 group induced steric hindrance during the self-polymerization because of its bulkier size than the methyl group and limited the amount of successful π-conjugations in the same time frame. Although further investigation will be needed to clarify the matter, the combination of the above assumptions is in agreement with the TGA results discussed below.
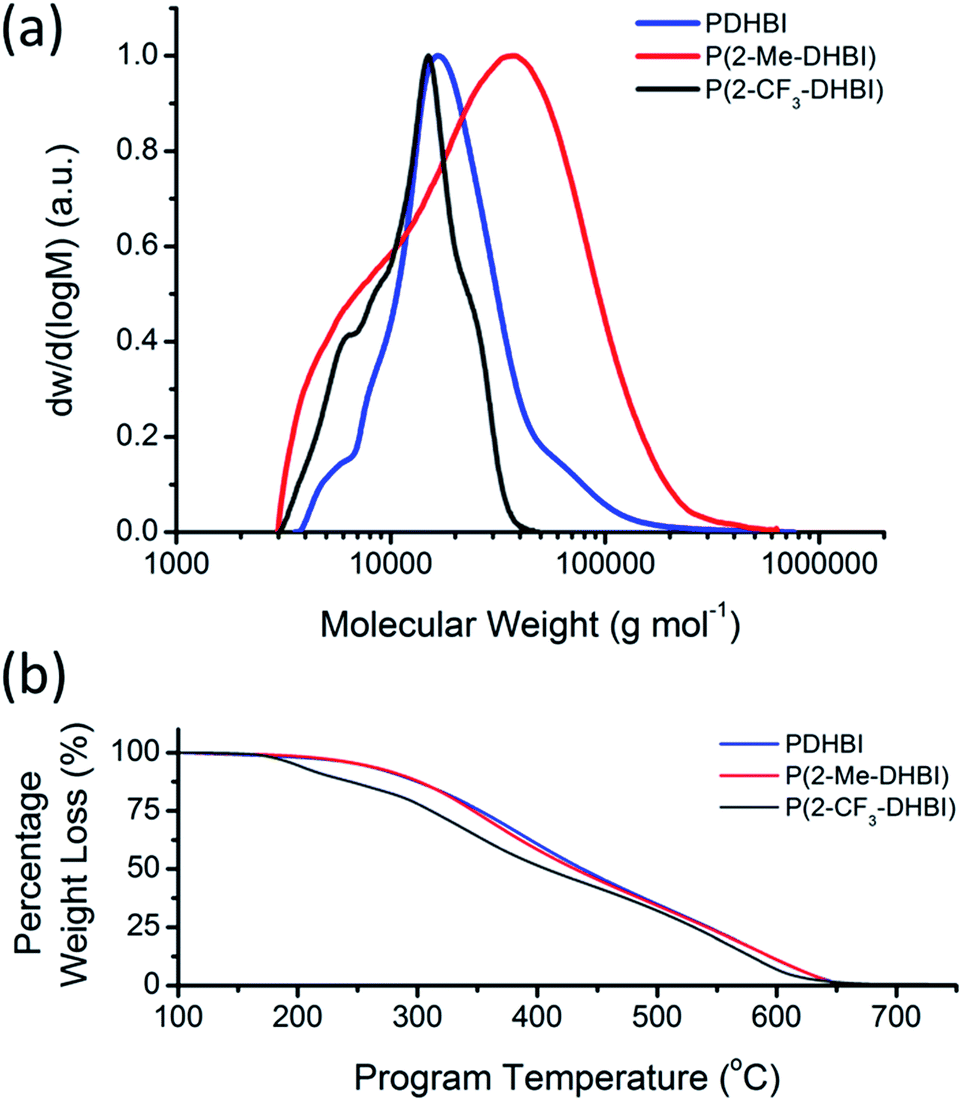 | ||
| Fig. 9 Molecular weight distribution (a) and thermal degradation of PDHBI, P(2-Me-DHBI) and P(2-CF3-DHBI) (b). | ||
Since the 2-substituted polymers are not cross-linked, no coated substrate has been prepared. However, TGA was performed on the free polymers to elucidate the respective thermal degradation in order to demonstrate the possibility to modify, in particular, the thermal properties by the substitution of different functional groups at the 2-carbon position. As illustrated by Fig. 9b, P(2-Me-DHBI) exhibited unexpectedly similar on-set degradation temperature and high-temperature resistance as PDHBI, though a small change in gradient around 400 °C which was believed to be related to the degradation of the methyl group. The high level of similarity suggests that the extent of cross-linking of PDHBI may not be enough to significantly strengthen the polymer structure, but the benzimidazole origin allowed P(2-Me-DHBI) to retain the resistance to high temperatures. In stark contrast, P(2-CF3-DHBI) exhibited less stability and the thermal degradation underwent several stages. As discussed previously, the CF3 group is bulkier than the methyl group whilst its electron-withdrawing effect can potentially disrupt the stability of the π-conjugation. As a result, the onset degradation of P(2-CF3-DHBI) was at a lower temperature (197 °C). The tunability of thermal properties reported here, in turn, demonstrated the versatility of the reaction between o-benzenediamine and carboxylic acids as a synthesis platform for preparing DHBI-based functionalized monomers. Although the substituent at C-2 impedes the establishment of linkages between monomer units, dopamine or DHBI can be employed as a comonomer to aid coating formation with considerable structural stability in a similar manner to the copolymerization between dopamine and the non-cross-linkable 5,6-dihydroxy-1H-indazole which have been previously demonstrated by our group.34 However, the ability of applying the 2-substituted monomers for surface coating and property modification was not the focus of this work. Such potential shall be further explored in future research, along with the possibility to employ diacids,63–65 or even triacids, to form linkages at C-2 prior to the polymerization to enforce a highly cross-linked structure for the resulting polymer. It gives the opportunity to develop polymeric materials with tailored performance, and perhaps comparable to that of PBIs, via the self-polymerization of DHBI-based monomers under mild aqueous conditions.
Conclusions
In conclusion, we have demonstrated the potential to perform aqueous-based redox-facilitated self-polymerization of DHBI and its 2-subtituted derivatives. This branch of chemistry is no longer exclusive to catecholamines and other polyphenol structures, but has been proven expandable to novel synthetic heterocycles. Although DHBI monomers share similar self-polymerization mechanism to dopamine, the reaction can proceed in a significantly faster manner under the same reaction conditions and can be further accelerated by UV-stimulation. In order to use PDHBI-based materials for a particular application, further modification may be required to enhance or tailor the performance. We showed that it is possible to manipulate the properties, e.g. thermal stability, readily by copolymerization with dopamine accredited to the compatibility of the reaction system. Moreover, property manipulation can also be achieved through selecting the desired functionality for the 2-carbon position of DHBI accredited to the versatile chemistry between o-benzenediamine and carboxylic acids, which has drastically increased the solubility of the polymers formed. This synthesis platform will become a new channel for generating a spectrum of monomers based on DHBI for the aqueous-based preparation of functional polymeric materials to suit specific applications.Author contributions
K. W. F., M. B. P., P. E., and A. M. G. performed experimental work and analyzed experimental results. K. W. F., M. B. P., and A. M. G. wrote the manuscript. A. M. G. conceived and designed the project.Conflict of interest
The authors declare no competing financial interests.Acknowledgements
We thank Dr James M. Hook and Dr Aditya Rawal for helpful discussions and aid in performing the solid-state NMR experiments. We also thank Dr Donald S. Thomas for aid in performing EPR experiments and the analysis of the results. The equipment in the NMR Facility at the Mark Wainwright Analytical Centre is supported by the Australian Research Council through Linkage Infrastructure, Equipment and Facilities Funding Scheme (LE0989541 and LE120100027) and the UNSW Major Research Equipment and Infrastructure Initiative (MREII). We acknowledge the time devoted by Mr Josef Goding for performing CV experiment. We also acknowledge the support of the Electron Microscopy Unit (EMU), Mark Wainwright Analytical Centre, UNSW. K. W. F. would like to acknowledge the support of the Australian Government for scholarship funding in the form of an Australian Postgraduate Award and a UNSW Engineering Research Award.Notes and references
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- A. Postma, Y. Yan, Y. Wang, A. N. Zelikin, E. Tjipto and F. Caruso, Chem. Mater., 2009, 21, 3042–3044 CrossRef CAS.
- B. Yu, J. Liu, S. Liu and F. Zhou, Chem. Commun., 2010, 46, 5900–5902 RSC.
- S. Hong, K. Y. Kim, H. J. Wook, S. Y. Park, K. D. Lee, D. Y. Lee and H. Lee, Nanomedicine, 2011, 6, 793–801 CrossRef CAS PubMed.
- C. J. Ochs, T. Hong, G. K. Such, J. Cui, A. Postma and F. Caruso, Chem. Mater., 2011, 23, 3141–3143 CrossRef CAS.
- B. Yu, D. A. Wang, Q. Ye, F. Zhou and W. Liu, Chem. Commun., 2009, 6789–6791 RSC.
- J. Cui, Y. Yan, G. K. Such, K. Liang, C. J. Ochs, A. Postma and F. Caruso, Biomacromolecules, 2012, 13, 2225–2228 CrossRef CAS PubMed.
- J. Cui, Y. Wang, A. Postma, J. Hao, L. Hosta-Rigau and F. Caruso, Adv. Funct. Mater., 2010, 20, 1625–1631 CrossRef CAS.
- Y. Liu, K. Ai, J. Liu, M. Deng, Y. He and L. Lu, Adv. Mater., 2013, 25, 1353–1359 CrossRef CAS PubMed.
- S. M. Kang, M.-H. Ryou, J. W. Choi and H. Lee, Chem. Mater., 2012, 24, 3481–3485 CrossRef CAS.
- M.-H. Ryou, Y. M. Lee, J.-K. Park and J. W. Choi, Adv. Mater., 2011, 23, 3066–3070 CrossRef CAS PubMed.
- J. H. Kim, M. Lee and C. B. Park, Angew. Chem., Int. Ed., 2014, 53, 6280 CrossRef.
- M. Kohri, H. Kohma, Y. Shinoda, M. Yamauchi, S. Yagai, T. Kojima, T. Taniguchi and K. Kishikawa, Polym. Chem., 2013, 4, 2696–2702 RSC.
- S. P. Le-Masurier, G. Gody, S. Perrier and A. M. Granville, Polym. Chem., 2014, 5, 2816–2823 RSC.
- B. D. McCloskey, H. B. Park, H. Ju, B. W. Rowe, D. J. Miller and B. D. Freeman, J. Membr. Sci., 2012, 413, 82–90 CrossRef.
- Q. Liu, N. Wang, J. Caro and A. Huang, J. Am. Chem. Soc., 2013, 135, 17679–17682 CrossRef CAS PubMed.
- I. You, S. M. Kang, S. Lee, Y. O. Cho, J. B. Kim, S. B. Lee, Y. S. Nam and H. Lee, Angew. Chem., Int. Ed., 2012, 51, 6126–6130 CrossRef CAS PubMed.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- J. Liebscher, R. Mrówczyński, H. A. Scheidt, C. Filip, N. D. Hădade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef CAS PubMed.
- M. d'Ischia, A. Napolitano and A. Pezzella, Eur. J. Org. Chem., 2011, 2011, 5501–5516 CrossRef.
- M. Ambrico, P. F. Ambrico, A. Cardone, N. F. Della Vecchia, T. Ligonzo, S. R. Cicco, M. M. Talamo, A. Napolitano, V. Augelli, G. M. Farinola and M. d'Ischia, J. Mater. Chem. C, 2013, 1, 1018–1028 RSC.
- M. Ambrico, N. F. D. Vecchia, P. F. Ambrico, A. Cardone, S. R. Cicco, T. Ligonzo, R. Avolio, A. Napolitano and M. d'Ischia, Adv. Funct. Mater., 2014, 24, 7161–7172 CAS.
- L. García-Fernández, J. Cui, C. Serrano, Z. Shafiq, R. A. Gropeanu, V. S. Miguel, J. I. Ramos, M. Wang, G. K. Auernhammer, S. Ritz, A. A. Golriz, R. Berger, M. Wagner and A. del Campo, Adv. Mater., 2013, 25, 529–533 CrossRef PubMed.
- Z. Shafiq, J. Cui, L. Pastor-Pérez, V. San Miguel, R. A. Gropeanu, C. Serrano and A. del Campo, Angew. Chem., 2012, 124, 4408–4411 CrossRef.
- S. Hong, J. Kim, Y. S. Na, J. Park, S. Kim, K. Singha, G.-I. Im, D.-K. Han, W. J. Kim and H. Lee, Angew. Chem., Int. Ed., 2013, 52, 9187–9191 CrossRef CAS PubMed.
- S. M. Kang, J. Rho, I. S. Choi, P. B. Messersmith and H. Lee, J. Am. Chem. Soc., 2009, 131, 13224–13225 CrossRef CAS PubMed.
- M. E. Lynge, R. v. d. Westen, A. Postma and B. Stadier, Nanoscale, 2011, 3, 4916–4928 RSC.
- P. Manini, M. d'Ischi and G. Prota, J. Org. Chem., 2001, 66, 5048–5053 CrossRef CAS PubMed.
- S. Hong, J. Yeom, I. T. Song, S. M. Kang, H. Lee and H. Lee, Adv. Mater. Interfaces, 2014, 1, 1400113 Search PubMed.
- A. Pezzella, A. Iadonisi, S. Valerio, L. Panzella, A. Napolitano, M. Adinolfi and M. d'Ischia, J. Am. Chem. Soc., 2009, 131, 15270–15275 CrossRef CAS PubMed.
- L. Capelli, O. Crescenzi, P. Manini, A. Pezzella, V. Barone and M. d'Ischia, J. Org. Chem., 2011, 76, 4457–4466 CrossRef CAS PubMed.
- M. B. Peterson, S. P. Le-Masurier, K. Lim, J. M. Hook, P. Martens and A. M. Granville, Macromol. Rapid Commun., 2014, 35, 291–297 CrossRef CAS PubMed.
- K. W. Fan, J. J. Roberts, P. J. Martens, M. H. Stenzel and A. M. Granville, J. Mater. Chem. B, 2015, 3, 7457–7465 RSC.
- Y. Yao, Y. Che and J. Zheng, Cryst. Growth Des., 2008, 8, 2299–2306 CAS.
- S. Kapitza, M. Pongratz, M. A. Jakupec, P. Heffeter, W. Berger, L. Lackinger, B. K. Keppler and B. Marian, J. Cancer Res. Clin. Oncol., 2005, 131, 101–110 CrossRef CAS PubMed.
- P. Renton, B. Green, S. Maddaford, S. Rakhit and J. S. Andrews, ACS Med. Chem. Lett., 2012, 3, 227–231 CrossRef CAS PubMed.
- P. Sudhesh, K. Tamilarasan, P. Arumugam and S. Berchmans, ACS Appl. Mater. Interfaces, 2013, 5, 8263–8266 CAS.
- L. R. Belohlav, Angew. Makromol. Chem., 1974, 40, 465–483 CrossRef.
- M. F. Flanagan and I. C. Escobar, J. Membr. Sci., 2013, 434, 85–92 CrossRef CAS.
- L. Liang, Q. Gan and P. Nancarrow, J. Membr. Sci., 2013, 450, 407–417 CrossRef.
- C.-H. Shen and S. L.-c. Hsu, J. Membr. Sci., 2013, 443, 138–143 CrossRef CAS.
- X. Li, G. Qian, X. Chen and B. C. Benicewicz, Fuel Cells, 2013, 13, 832–842 CAS.
- Y. Wang, Z. Shi, J. Fang, H. Xu and J. Yin, Carbon, 2011, 49, 1199–1207 CrossRef CAS.
- Y. Wang, J. Yu, L. Chen, Z. Hu, Z. Shi and J. Zhu, RSC Adv., 2013, 3, 20353–20362 RSC.
- L. Weinberger and A. R. Day, J. Org. Chem., 1959, 24, 1451–1455 CrossRef CAS.
- A. O. H. El-Nezhawy, A. R. Biuomy, F. S. Hassan, A. K. Ismaiel and H. A. Omar, Bioorg. Med. Chem., 2013, 21, 1661–1670 CrossRef CAS PubMed.
- O. René, A. Souverneva, S. R. Magnuson and B. P. Fauber, Tetrahedron Lett., 2013, 54, 201–204 CrossRef.
- H. M. Alkahtani, A. Y. Abbas and S. Wang, Bioorg. Med. Chem. Lett., 2012, 22, 1317–1321 CrossRef CAS PubMed.
- W. Sheng, B. Li, X. Wang, B. Dai, B. Yu, X. Jia and F. Zhou, Chem. Sci., 2015, 6, 2068–2073 RSC.
- Z. Bo, W. Li, R. Qin, C. Li, C. Du and Z. Liu, Preparation of conjugated polymer, CN 101875716 A, 2010, p. 12.
- F. Li, B. Frett and H.-y. Li, Synlett, 2014, 25, 1403–1408 CrossRef PubMed.
- M. D'Ischia, O. Crescenzi, A. Pezzella, M. Arzillo, L. Panzella, A. Napolitano and V. Barone, Photochem. Photobiol., 2008, 84, 600–607 CrossRef PubMed.
- H. Okuda, K. Wakamatsu, S. Ito and T. Sota, J. Phys. Chem. A, 2008, 112, 11213–11222 CrossRef CAS PubMed.
- C. A. Matuszak and A. J. Matuszak, J. Chem. Educ., 1976, 53, 280 CrossRef CAS.
- R. T. Borchardt and P. Bhatia, J. Med. Chem., 1982, 25, 263–271 CrossRef CAS PubMed.
- S. Jens-Uwe and J. S. B. David, Methods Appl. Fluoresc., 2014, 2, 024005 CrossRef.
- K. Y. Ju, Y. Lee, S. Lee, S. B. Park and J. K. Lee, Biomacromolecules, 2011, 12, 625–632 CrossRef CAS PubMed.
- M. d'Ischia, A. Napolitano, A. Pezzella, P. Meredith and T. Sarna, Angew. Chem., Int. Ed., 2009, 48, 3914–3921 CrossRef PubMed.
- N. F. della Vecchia, P. Cerruti, G. Gentile, M. E. Errico, V. Ambrogi, G. D'Errico, S. Longobardi, A. Napolitano, L. Paduano, C. Carfagna and M. d'Ischia, Biomacromolecules, 2014, 15, 3811–3816 CrossRef CAS PubMed.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- Z. Y. Xi, Y. Y. Xu, L. P. Zhu, Y. Wang and B. K. Zhu, J. Membr. Sci., 2009, 327, 244–253 CrossRef CAS.
- L. Li and G. J. Clarkson, Org. Lett., 2007, 9, 497–500 CrossRef CAS PubMed.
- A. Herbst, C. Bronner, P. Dechambenoit and O. S. Wenger, Organometallics, 2013, 32, 1807–1814 CrossRef CAS.
- Y.-H. So and J. P. Heeschen, J. Org. Chem., 1997, 62, 3552–3561 CrossRef CAS.
- L. Zhang, S. Roy, Y. Chen, E. K. Chua, K. Y. See, X. Hu and M. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 18644–18652 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C, 2D NMR spectra, 13C solid-state NMR spectra, ATR-FTIR spectra, TGA thermograms, data obtained from DLS and GPC. See DOI: 10.1039/c5ra25590b |
| ‡ The coating thickness of 1 and 3 h PDA kinetics samples could not be determined accurately due to extensive aggregation. |
| This journal is © The Royal Society of Chemistry 2016 |