
Energy-efficient degradation of rhodamine B in a LED illuminated photocatalytic fuel cell with anodic Ag/AgCl/GO and cathodic ZnIn2S4 catalysts†
Abstract
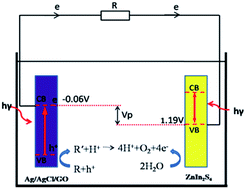
Photocatalytic fuel cells (PFCs) are a newly developed technology that degrade pollutants and simultaneously generate electricity. Stainless steel electrodes loaded with anodic Ag/AgCl/GO and cathodic ZnIn2S4, formed a one-chambered PFC, in which rhodamine B (RhB) was degraded under visible light (2 W LED). After 1 h of irradiation, 87.4% of the RhB was degraded and a 0.52 mA cm−2 current density was generated when the external resistance was 1 Ω. Increasing this resistance lowered the current density and decreased degradation. The current and cell voltage are affected by the degradation efficiency over the electrodes, and the photocatalytic electrode with higher degradation activity functioned as the anode, because of its relatively richer supply of electrons compared to the other. Ag/AgCl/GO can function as a cathode in a two-chambered PFC reactor with Fe as anode, which also had high degradation efficiency and higher electricity generation performance. The characteristics of the photoelectrodes were investigated using scanning or transparent electronic microscopy (SEM, TEM), continuous cyclic voltammograms (CV) and Electrochemical Impedance Spectroscopy (EIS). Electron Spin Resonance (ESR) was used to detect the reactive oxygen species (ROS). The effect of pH and RhB concentration on the degradation performance of this PFC was investigated. The PFCs can work in a broad range of pH.
 Please wait while we load your content...
Please wait while we load your content...

