
Design, synthesis and RAFT polymerisation of a quinoline-based monomer for use in metal-binding composite microfibers†
Abstract
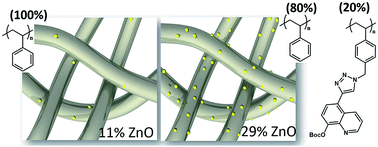
Metal-binding polymer fibres have attracted major attention for diverse applications in membranes for metal sequestration from waste waters, non-woven wound dressings, matrices for photocatalysis, and many more. This paper reports the design and synthesis of an 8-hydroxyquinoline-based zinc-binding styrenic monomer, QuiBoc. Its subsequent polymerisation by reversible addition–fragmentation chain transfer (RAFT) yielded well-defined polymers, PQuiBoc, of controllable molar masses (6 and 12 kg mol−1) with low dispersities (Đ, Mw/Mn < 1.3). Protected (PQuiBoc) and deprotected (PQuiOH) derivatives of the polymer exhibited a high zinc-binding capacity, as determined by semi-quantitative SEM/EDXA analyses, allowing the electrospinning of microfibres from a PQuiBoc/polystyrene (PS) blend without the need for removal of the protecting group. Simple “dip-coating” of the fibrous mats into ZnO suspensions showed that PQuiBoc/PS microfibres with only 20% PQuiBoc content had almost three-fold higher loadings of ZnO (29%) in comparison to neat PS microfibres (11%).
 Please wait while we load your content...
Please wait while we load your content...

