
Modulating hierarchical self-assembly behavior of a peptide amphiphile/nonionic surfactant mixed system
Abstract
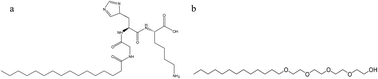
The self-assembly behavior of a nonionic surfactant (n-dodecyl tetraethylene monoether, C12E4) and a peptide amphiphile (PA, C16-GK-3) mixed system was investigated using a combination of microscopic, scattering and spectroscopic techniques, including transmission electron microscopy (TEM), field emission-scanning electron microscopy (FE-SEM), atomic force microscopy (AFM), polarized optical microscopy (POM) observations, small-angle X-ray scattering (SAXS), Fourier transform infrared (FT-IR) spectroscopy, circular dichroism (CD) and rheological measurements. The change of the contents of C16-GK-3 and C12E4 induced the transitions in the nanostructures and simultaneously led to changes in macroscopic properties, i.e., mixtures of C12E4 with C16-GK-3 can be hierarchically self-assembled into various helical nanofibers and then further assembled to dandelion-like and dendrite nanostructures by changing the content of C16-GK-3 and C12E4, which resulted in transitions from solution to two phase, sol and hydrogel states that were noted on increasing the concentration of C16-GK-3 at a fixed concentration of C12E4 or varying C12E4 concentration at a fixed concentration of C16-GK-3. On the basis of a series of characterizations, we proposed a possible mechanism of the self-assembly, for which the hydrogen bonding interaction between the headgroups of C16-GK-3 and between C16-GK-3 and C12E4, as well as hydrophobic interaction between the alkyl chains of C16-GK-3 and C12E4, were the main driving forces.
 Please wait while we load your content...
Please wait while we load your content...

