
Intracellular thiol-responsive nanosized drug carriers self-assembled by poly(ethylene glycol)-b-poly(ε-caprolactone)-b-poly(ethylene glycol) having multiple bioreducible disulfide linkages in hydrophobic blocks†
Seung Yeon Moon‡
a,
Yeon Su Choi‡a,
Jung-Kyo Choa,
Minjong Yub,
Eunji Leeb,
Kang Moo Huhc,
Don Haeng Leede,
Jong-Ho Kim*f and
Han Chang Kang*a
aDepartment of Pharmacy and Integrated Research Institute of Pharmaceutical Sciences, College of Pharmacy, The Catholic University of Korea, 43 Jibong-ro, Wonmi-gu, Bucheon-si, Gyeonggi-do 14662, Republic of Korea. E-mail: hckang@catholic.ac.kr; Fax: +82-2-2164-4059; Tel: +82-2-2164-6533
bGraduate School of Analytical Science and Technology, Chungnam National University, 99 Daehak-ro, Yuseong-gu, Daejeon 34134, Republic of Korea
cDepartment of Polymer Science and Engineering, Chungnam National University, 99 Daehak-ro, Yuseong-gu, Daejeon 34134, Republic of Korea
dDivision of Gastroenterology and Hepatology, Department of Internal Medicine, Inha University Hospital, 27 Inhang-ro, Jung-gu, Incheon 22332, Republic of Korea
eUtah-Inha Drug Delivery Systems and Advanced Therapeutics Research Center, 9 Songdomirae-ro, Yeonsu-gu, Incheon 21988, Republic of Korea
fDepartment of Pharmaceutical Sciences, Kyung Hee University, 26 Kyungheedae-ro, Dongdaemoon-gu, Seoul 02447, Republic of Korea. E-mail: jonghokim@khu.ac.kr; Fax: +82-2-966-3885; Tel: +82-2-961-9312
First published on 26th January 2016
Abstract
To achieve effective intracellular drug release from nanostructures, we designed high molecular weight (HMW) reducible poly(ε-caprolactone) (PCL) structures synthesized by a coupling reaction between the low molecular weight (LMW) PCL diol and a single disulfide-containing dicarboxylic acid. The synthesized HMW-reducible PCL (RSPCL) polymers with approximately 7 disulfide bonds were further linked with methoxy poly(ethylene glycol) (mPEG), resulting in mPEG-b-RSPCL-b-mPEG (mPEG-RSPCL) copolymers. In aqueous environments, the mPEG-RSPCL copolymers were self-assembled to construct nanoparticles (NPs) of less than 100 nm with nearly neutral zeta-potentials, and the NPs had negligible cytotoxicity below 0.2 mg mL−1. In a thiol-rich environment (10 mM), mPEG-RSPCL NPs increased in size over time, and their polymer components completely degraded to mPEG and LMW PCL derivatives. The mPEG-RSPCL NPs were able to load both water-soluble doxorubicin hydrochloride (DOX·HCl) and water-insoluble doxorubicin (DOX). Based on different cellular uptakes of free drugs and drug-loaded NPs in HeLa and HepG2 cells, DOX·HCl-loaded mPEG-RSPCL NPs showed approximately 1.7–3.6-fold and 20-fold higher anti-tumor effects than free DOX·HCl and DOX-loaded mPEG-RSPCL NPs, respectively. Additionally, DOX-mPEG-RSPCL NPs represented similar or lower drug efficacy than free DOX. Especially, DOX·HCl-loaded mPEG-RSPCL NPs represented superior thiol-triggered drug release and killing effects than their control NPs having fewer or no disulfide bonds. In conclusion, the designed mPEG-RSPCL NPs are potentially useful as nanosized drug carriers for the effective intracellular release of hydrophilic and hydrophobic drugs in the cytosol and/or nucleus.
Introduction
In aqueous environments, amphiphilic copolymers composed of hydrophilic and hydrophobic components can be spontaneously self-assembled to form various nanostructures (e.g., spherical micelle, cylindrical micelle, planar bilayer, polymersome, reverse micelle) at certain ratios (e.g., weight, volume, etc.) of hydrophilic and hydrophobic blocks.1–3 Micellar nanostructures have hydrophobic cores for loading hydrophobic chemical drugs, whereas polymersomes can encapsulate both hydrophobic and hydrophilic payloads in the hydrophobic compartment of a bilayer shell and in the aqueous interior separated from the outside by the bilayer shell, respectively.4–7 These varieties of nanostructures and loadable drugs have stimulated a continuously growing interest in amphiphilic copolymers and their drug delivery applications.Various functions, such as biocompatibility, biodegradation, and stimuli-triggered drug release, have been introduced into amphiphilic copolymers to enhance their clinical availability and drug efficacy.8 In most amphiphilic copolymers, poly(ethylene glycol) (PEG) has been utilized as a hydrophilic component because PEG represents excellent biocompatibility, including non-toxicity, non-immunogenicity, non-antigenicity, and non-tumorigenesis, and it prevents non-specific interactions with blood components that trigger protein adsorption, blood coagulation, and complement activation.9,10 Additionally, polyesters such as poly(glycolic acid) (PGA), poly(ε-caprolactone) (PCL), poly(lactic acid) (PLA), and poly(lactic-co-glycolic acid) (PLGA) have been extensively utilized as hydrophobic blocks because the polymers can be degraded into small acid metabolites, carbon dioxide, and water, which exist in the body, via hydrolysis and enzymatic biodegradation, resulting in excellent biocompatibility.3,11,12 Thus, PEG-polyester block copolymers having various molecular architectures (e.g., grafted, branched, star-shaped, linear) and molecular weights have been synthesized and used to tune nanostructures such as spherical micelles, worm-like micelles, and vesicles for delivery of chemical and biological therapeutics.3,11–14
PEG-polyester nanostructures are destabilized by slow hydrolysis-induced biodegradation of hydrophobic polyester blocks. However, their nanosized drug carriers do not provide quick release or intracellular stimulus-triggered release of payloads. Thus, certain chemical bonds and/or components responding to intracellular stimuli such as pH, enzymes, and glutathione (GSH) have been inserted into PEG-polyester copolymers to promote quick drug release in intracellular environments.15–19 However, an acidic pH as a trigger may not clearly distinguish intracellular pH (i.e., endolysosomal pH of 5–7) from extracellular pH (i.e., pH 6.5–7) in pathological environments such as solid tumors and inflammation sites.20–24 The use of enzymes that exist primarily in lysosomes can result in therapeutic loss via enzymatic damage of payloads before lysosomal escape.22 Recently, many researchers have focused on the disulfide–thiol exchange reaction19,25–31 because extracellular and cytosolic and nuclear concentrations of GSH, a biological thiol, range from 0.02–2 μM and 1–11 mM, respectively.18,26 Unlike pH and enzymes, the distinguishable difference of GSH levels has contributed to stable nanocarriers with minimal drug loss in extracellular environments but has destabilized them for quick drug release in the cytosol and the nucleus.
The locations of disulfide bonds have mostly been in connecting points between the hydrophilic blocks and hydrophobic blocks in PEG-polyester copolymers32,33 and crosslinks in chemically crosslinked hydrophobic cores in PEG-polyester nanoparticles.34–36 Zhong et al. designed PEG-ss-PCL diblock copolymers with a single disulfide bond located at the bridging point between two polymer blocks. Their non-toxic reduction-sensitive micelles showed faster release of hydrophobic doxorubicin (DOX) than the reduction-insensitive counterpart.33 Recently, Kumar et al., designed PCL-ss-PCL which was further reacted with PEG methacrylate (PEGMA) to synthesized PEGMA-PCL-ss-PCL-PEGMA triblock copolymers. Although the triblock copolymers were not compared with other non-reducible or reducible control polymeric systems, one disulfide bond located in the center of hydrophobic PCL blocks induced GSH-triggered drug release from their nanoparticles.37 In addition, although polyesters as disulfide-containing hydrophobic blocks were not used, more hydrophobic block of poly(disulfide) (PDS) in poly(triethylene glycol monomethyl ether) methacrylate-b-PDS-b-poly(triethylene glycol monomethyl ether)methacrylate (PTEGMA-b-PDS-b-PTEGMA) triblock copolymers caused slower drug release from their micelles responding to concentration of GSH.38
In this study, the amphiphilic block copolymers composed of PEG and PCL were selected and contained reduction-responsive disulfide bonds. However, unlike the single disulfide bond in PEG-ss-PCL diblock copolymers, multiple disulfide bonds were located at two linking points between the PEG block and the high molecular weight (HMW) reducible PCL (RSPCL) block as well as in several connecting points among low molecular weight (LMW) PCL blocks because the HMW RSPCL block was prepared by connecting several LMW PCL blocks with disulfide-containing molecules (Fig. 1). It is expected that PEG-RSPCL copolymers with multiple disulfide bonds will have an accelerated release of payloads in their nanostructures when exposed to the thiol-rich cytosol or nucleus. Thus, the nanostructures of methoxy poly(ethylene glycol)-b-reducible poly(ε-caprolactone)-b-methoxy poly(ethylene glycol) (mPEG-b-RSPCL-b-mPEG) copolymers with multiple disulfide bonds were evaluated for size, zeta-potential, thiol-induced degradation and size change, morphology, and in vitro cytotoxicity. After loading either a hydrophobic DOX or its salt form (i.e., a hydrophilic DOX hydrochloride (DOX·HCl)) into mPEG-RSPCL-based nanoparticles (NPs), their physicochemical and biological characteristics were investigated for particle size, zeta-potential, morphology, drug loading capacity, drug release, in vitro anti-tumor effects, in vitro cellular uptake, and intracellular drug distribution.
 | ||
| Fig. 1 (a) Preparation schemes of mPEG-RSPCL, mPEG-ssPCL, and mPEG-PCL copolymers and their drug-loaded nanoparticles and (b) design concept of drug-loaded mPEG-RSPCL nanoparticles in cells. | ||
Results and discussion
Synthesis and chemical and physical characteristics of mPEG-RSPCL copolymers
As shown in Fig. 2(a), the mPEG-RSPCL copolymers were synthesized in two steps. In the first step, HMW RSPCL polymers were synthesized by a conventional conjugation between two carboxylic acids of single disulfide bond-containing 3,3′-dithiodipropionic acid (DTPA) and two hydroxyl groups of LMW PCL diol (MW 530 Da and 1250 Da) in the presence of N,N′-dicyclohexylcarbodiimide (DCC), 4-(dimethylamino)pyridine (DMAP), and triethylamine (TEA). The next step was a coupling reaction between one or two carboxylic acids at the ends of the synthesized HMW RSPCL polymers and one or two mPEG molecules with one hydroxyl group, resulting in the formation of mPEG-RSPCL copolymers, including mPEG-b-RSPCL-b-mPEG triblock copolymers and mPEG-b-RSPCL diblock copolymers. In addition, mPEG-b-ssPCLss-b-mPEG (mPEG-ssPCL) and mPEG-b-PCL-b-mPEG copolymers as control polymers were synthesized as shown in Fig. 2(b). | ||
| Fig. 2 Synthetic scheme of (a) the designed mPEG-RSPCL copolymers and (b) the control mPEG-ssPCL and mPEG-PCL copolymers. | ||
To confirm the chemical structures and MWs of the synthesized RSPCL polymers and mPEG-RSPCL copolymers, the polymers were analyzed by 1H-NMR in CDCl3; the peak assignments are represented in Fig. 3(a) for RSPCL0.5 polymers and mPEG-RSPCL0.5 copolymers and Fig. S1† for RSPCL1.2 polymers and mPEG-RSPCL1.2 copolymers. The major peaks of the synthesized RSPCL0.5 polymers and RSPCL1.2 polymers were observed at δ 3.7 ppm of the PCL backbone and δ 2.7 ppm of DTPA, indicating the synthesis of RSPCL polymers. Based on the integration ratio between peaks representing –OC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CH2CH2SSCH2CH2C(O)O– in DTPA and –C(O)OCH2CH2OCH2CH2OC(O)– in the PCL backbone, the average numbers of disulfide bonds in the RSPCL0.5 and RSPCL1.2 polymers were 7 and 7.5, respectively, and the average numbers of the LMW PCL0.5 in HMW RSPCL0.5 polymers and PCL1.2 blocks in RSPCL1.2 polymers were 6 and 6.5, respectively. The estimated MWs of RSPCL0.5 and RSPCL1.2 polymers were 5130 and 9310 Da, respectively.
O)CH2CH2SSCH2CH2C(O)O– in DTPA and –C(O)OCH2CH2OCH2CH2OC(O)– in the PCL backbone, the average numbers of disulfide bonds in the RSPCL0.5 and RSPCL1.2 polymers were 7 and 7.5, respectively, and the average numbers of the LMW PCL0.5 in HMW RSPCL0.5 polymers and PCL1.2 blocks in RSPCL1.2 polymers were 6 and 6.5, respectively. The estimated MWs of RSPCL0.5 and RSPCL1.2 polymers were 5130 and 9310 Da, respectively.
 | ||
| Fig. 3 (a) 1H-NMR spectra and (b) GPC chromatograms of the synthesized mPEG-RSPCL0.5 copolymers. | ||
With the attachment of mPEG at two ends of the RSPCL polymers, newly introduced peaks due to mPEG were observed at δ 4.23 ppm, δ 3.69 ppm, δ 3.64 ppm, and δ 3.38 ppm in the mPEG-RSPCL0.5 and mPEG-RSPCL1.2 copolymers (Fig. 3(a) and S1(a),† respectively). Based on the integration ratio between the peaks representing –OCH3 in mPEG and –C(O)OCH2CH2OCH2CH2OC(O)– in the RSPCL backbone, the average number of mPEG per RSPCL0.5 backbone in mPEG-RSPCL0.5 copolymers and of mPEG per RSPCL1.2 backbone in mPEG-RSPCL1.2 copolymers was 1.8 and 1.7, respectively. The estimated MWs of the mPEG-RSPCL0.5 and mPEG-RSPCL1.2 copolymers were 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 270 and 19830 Da, respectively. The estimated physical properties of the synthesized RSPCL polymers and mPEG-RSPCL copolymers according to the 1H-NMR data are summarized in Table 1.
270 and 19830 Da, respectively. The estimated physical properties of the synthesized RSPCL polymers and mPEG-RSPCL copolymers according to the 1H-NMR data are summarized in Table 1.
| In feed | In synthesis | ||||||
|---|---|---|---|---|---|---|---|
| Mn of mPEGa | Mn of PCLa | Mnb | Mnc | PDIc | # of disulfide bonds in polymerb | # of mPEG chains in polymerb | |
| a Reported by the manufacturer.b Estimated from 1H-NMR spectra.c Determined by GPC. | |||||||
| PCL0.5 diol | — | 530 Da | 610 Da | 900 Da | 1.26 | — | — |
| PCL1.2 diol | — | 1250 Da | 1190 Da | 2330 Da | 1.26 | — | — |
| PCL5 diol | — | 5000 Da | 5120 Da | 7860 Da | 1.68 | — | — |
| mPEG | 5000 Da | — | 6190 Da | 8350 Da | 1.04 | — | — |
| RSPCL0.5 | — | 530 Da | 5130 Da | 4890 Da | 1.51 | 7 | — |
| RSPCL1.2 | — | 1250 Da | 9310 Da | 11880 Da |
1.76 | 7.5 | — |
| ssPCL5ss | — | 5000 Da | 5390 Da | 8059 Da | 1.58 | 2 | — |
| mPEG-RSPCL0.5 | 5000 Da | 530 Da | 16270 Da |
18590 Da |
1.17 | 7 | 1.8 |
| mPEG-RSPCL1.2 | 5000 Da | 1250 Da | 19830 Da |
28620 Da |
1.18 | 7.5 | 1.7 |
| mPEG-PCL5 | 5000 Da | 5000 Da | 14880 Da |
16120 Da |
1.7 | — | 1.9 |
| mPEG-ssPCL5 | 5000 Da | 5000 Da | 16220 Da |
18120 Da |
1.77 | 1.8 | 1.8 |
Further analysis of the synthesized RSPCL polymers and mPEG-RSPCL copolymers was performed by GPC because the synthesized mPEG-RSPCL copolymers and a mixture of mPEG and RSPCL polymers were not clearly distinguished by the previous NMR results. As shown in Fig. 3(b) and S2(a),† the elution times of mPEG-RSPCL copolymers were earlier than those of the RSPCL polymers, indicating that mPEG blocks were chemically linked with RSPCL polymers. The shifted elution times corresponded with increasing MW, with as much as 13700 Da for mPEG-RSPCL0.5 copolymers and 16740 Da for mPEG-RSPCL1.2 copolymers, indicating that the number of mPEG molecules in mPEG-RSPCL0.5 and mPEG-RSPCL1.2 copolymers were approximately 1.64 and 2.0, respectively. The MWs and PDIs of the synthesized RSPCL polymers and mPEG-RSPCL copolymers are summarized in Table 1. According to the 1H-NMR and GPC results, the polymeric architectures of the synthesized RSPCL polymers and mPEG-RSPCL copolymers were confirmed and seem to be almost mPEG-b-RSPCL-b-mPEG triblock copolymers.
In addition, mPEG-b-PCL5-b-mPEG (mPEG-PCL5) triblock copolymers and mPEG-ssPCL5ss-mPEG (mPEG-ssPCL5) triblock copolymer as control polymers (Fig. 1(a)) were synthesized as shown in Fig. 2(b) and their physical characteristics were summarized in Table 1 to compare with various characteristics of mPEG-RSPCL0.5 triblock copolymers. MWs of mPEG-PCL5 and mPEG-ssPCL5 copolymers were 14880 Da (16120 Da) and 16220 Da (18120 Da), respectively, based on 1H-NMR (GPC) spectra (Fig. S1(b), S1(c), S2(b), and S2(c)†) were similar to that of mPEG-RSPCL0.5 copolymers. Their molecular weights and 1H-NMR analysis indicated that the control polymers were triblock-shaped with mPEG blocks at two ends.
Preparation and physicochemical, degradation, and biological characteristics of the self-assembled mPEG-RSPCL NPs
The synthesized mPEG-RSPCL copolymers were self-assembled to construct NPs in aqueous environments, and the formed NPs were purified by a dialysis. The mean sizes of mPEG-RSPCL0.5 and mPEG-RSPCL1.2 NPs were approximately 87 and 63 nm, respectively, and the mean zeta-potentials were approximately −0.5 and −1.9 mV, respectively (Table 2). These size data indicate that the designed amphiphilic triblock mPEG-RSPCL copolymers could form self-assembled nanostructures smaller than 100 nm and with a narrow size distribution. Additionally, the almost neutral zeta-potentials suggest that mPEG may be well placed on the shell of the mPEG-RSPCL NPs. Two mPEG-PCL5 NPs and mPEG-ssPCL5 NPs as control NPs also showed approximately 39 nm and 51 nm in size, respectively and −0.16 ± 0.28 mV and −0.3 ± 0.40 mV in zeta-potential, respectively.| Code name of nanoparticle (NP) | Size (nm) | Zeta-potential (mV) | Targeted DLC (wt%) | Actual DLC (wt%) | Actual DLE (%) | Drug used | Drug-loading method |
|---|---|---|---|---|---|---|---|
| mPEG-RSPCL0.5 NP | 87 ± 19 | −0.5 ± 0.4 | — | — | — | — | — |
| mPEG-RSPCL1.2 NP | 63 ± 15 | −1.9 ± 0.3 | — | — | — | — | — |
| DOX-mPEG-RSPCL0.5 NP | 79 ± 15 | 1.2 ± 1.7 | 10 | 5.4 ± 0.2 | 54 ± 1.8 | DOX | Co-solvent dispersion (CD) |
| DOX-mPEG-RSPCL1.2 NP | 89 ± 18 | 0.3 ± 1.6 | 10 | 5.2 ± 0.5 | 52 ± 5.2 | DOX | CD |
| DOX·HCl-mPEG-RSPCL0.5 NP | 117 ± 21 | 1.4 ± 5.8 | 10 | 4.5 ± 0.1 | 45 ± 1.3 | DOX·HCl | Ammonium sulfate (AS) gradient |
| DOX·HCl-mPEG-RSPCL1.2 NP | 98 ± 20 | −1.7 ± 1.3 | 10 | 4.2 ± 0.6 | 42 ± 6.4 | DOX·HCl | AS gradient |
The mPEG-RSPCL NPs were designed to be degraded in a thiol-rich condition for their applications in intracellular drug delivery because it is known that the cytosolic and nuclear concentration of GSH, a thiol chemical in the body, are 50- to 1000-fold higher than extracellular concentrations.18,26 Thus, DTT (10 mM), a model thiol, was used to treat the designed NPs in DPBS at 37 °C, and the change in MW was monitored by GPC. As shown in Fig. 4(a), the average MWs (Mns) of mPEG-RSPCL0.5 and mPEG-RSPCL1.2 copolymers were 18590 and 28620 Da, respectively, before DTT (10 mM) treatment. DTT treatment reduced the average Mns to 1810 and 3870 Da, respectively, for 0.5 h post-treatment. More treatment time did not significantly change the MW. The average MWs of the polymer fragments were affected by MWs of mPEG and LMW PCL blocks. That is, as shown in Fig. 4(b) and S3,† mPEG blocks were detected at approximately 7.1 min of the elution time, whereas PCL0.5 and PCL1.2 derivatives were observed at 8.2 and 7.8 min, respectively. The DTT-induced degradation results indicate that disulfide bonds exist in the synthesized mPEG-RSPCL copolymers, that DTT (10 mM) treatment for only 30 min fully degraded all the disulfide bonds in mPEG-RSPCL copolymers, and that the copolymers could be degraded in thiol-rich intracellular environments.
 | ||
| Fig. 4 Thiol-induced MW change of mPEG-RSPCL copolymers in drug-free mPEG-RSPCL NPs: (a) time-dependent MW change of components in mPEG-RSPCL0.5 and mPEG-RSPCL1.2 NPs and (b) time-dependent GPC chromatograms of components in mPEG-RSPCL0.5 NPs. The degradation experiment was carried out at 37 °C. ([mPEG-RSPCL NP] = 0.25 mg mL−1, [DTT] = 10 mM). | ||
Although a thiol-triggered degradation of the designed mPEG-RSPCL copolymers in mPEG-RSPCL NPs was shown in DPBS (Fig. 4 and S3†), it is important to determine if the NPs maintain their physicochemical characteristics (e.g., size). Thus, their size changes were measured because the breakage of disulfide bonds in disulfide bond-containing NPs may result in either a size increase (due to interparticle aggregation among exposed hydrophobic parts) or decrease (due to loss of amphiphilicity in amphiphilic copolymers forming their nanostructures) with time. As shown in Fig. 5(a), in the absence of DTT, mPEG-PCL5 NPs, mPEG-ssPCL5 NPs, mPEG-RSPCL0.5 NPs, and mPEG-RSPCL1.2 NPs had less than 100 nm in number average diameter (around 100 nm in intensity average diameter as shown in Fig. S4†). However, although the size of mPEG-PCL5 NPs was not affected by the presence of DTT, DTT treatment (10 mM) caused the sizes of mPEG-ssPCL5 NPs, mPEG-RSPCL0.5 NPs, and mPEG-RSPCL1.2 NPs to gradually increase with increasing time (Fig. 5(a) and S4†). Interestingly, DTT induced a faster number-average size increase of mPEG-RSPCL1.2 NPs than mPEG-RSPCL0.5 NPs (i.e., the size change from approximately 90 to 200 nm in mPEG-RSPCL0.5 NPs versus the change from approximately 60 to 1000 nm in mPEG-RSPCL1.2 NPs) (Fig. 5(b)). The results may be due to both the number of disulfide bonds and the MW of the PCL diol. First, at a fixed concentration (i.e., mg mL−1) of mPEG-RSPCL NPs, the total number of disulfide bonds in mPEG-RSPCL0.5 NPs is greater than that of mPEG-RSPCL1.2 NPs because the former has a lower MW than the latter (Table 1). This indicates that a fixed concentration of DTT (10 mM) may be relatively higher for breaking disulfide bonds in mPEG-RSPCL1.2 NPs than in mPEG-RSPCL0.5 NPs, thus faster degradation occurs in the former. Really, higher concentrations of DTT caused the formation of larger aggregates from mPEG-RSPCL0.5 NPs (Fig. S5†). In addition, low DTT levels (i.e., <0.1 mM such as extracellular thiol concentrations) did not induce the aggregation of mPEG-RSPCL0.5 NPs, suggesting that mPEG-RSPCL0.5 NPs could be not degradable in the extracellular environments unlike the intracellular environments (Fig. S5†). Second, when degrading mPEG-RSPCL NPs in the presence of DTT, the degraded RSPCL fragments were similar to the LMW PCL used for synthesizing RSPCL as shown in Fig. 4(b) and S3.† That is, the degraded PCL1.2 derivatives from mPEG-RSPCL1.2 NPs were more hydrophobic and more water-insoluble than the degraded PCL0.5 derivatives from mPEG-RSPCL0.5 NPs, resulting in a hydrophobic interaction-induced interparticle aggregation and PCL fragment-mediated intermolecular aggregation due to the exposure of hydrophobic surfaces and components. Thus, the faster cleavage of disulfide bonds in mPEG-RSPCL1.2 NPs than in mPEG-RSPCL0.5 NPs could accelerate and increase the exposure of hydrophobic parts on the surface of the NPs, resulting in larger mPEG-RSPCL1.2 NPs than mPEG-RSPCL0.5 NPs. For mPEG-ssPCL5 NPs, DTT treatment caused quick cleavage of disulfide bonds and aggregated PCL5 derivatives, resulting that some particles were detected around 600 nm (Fig. 5(a)) but some were precipitated.
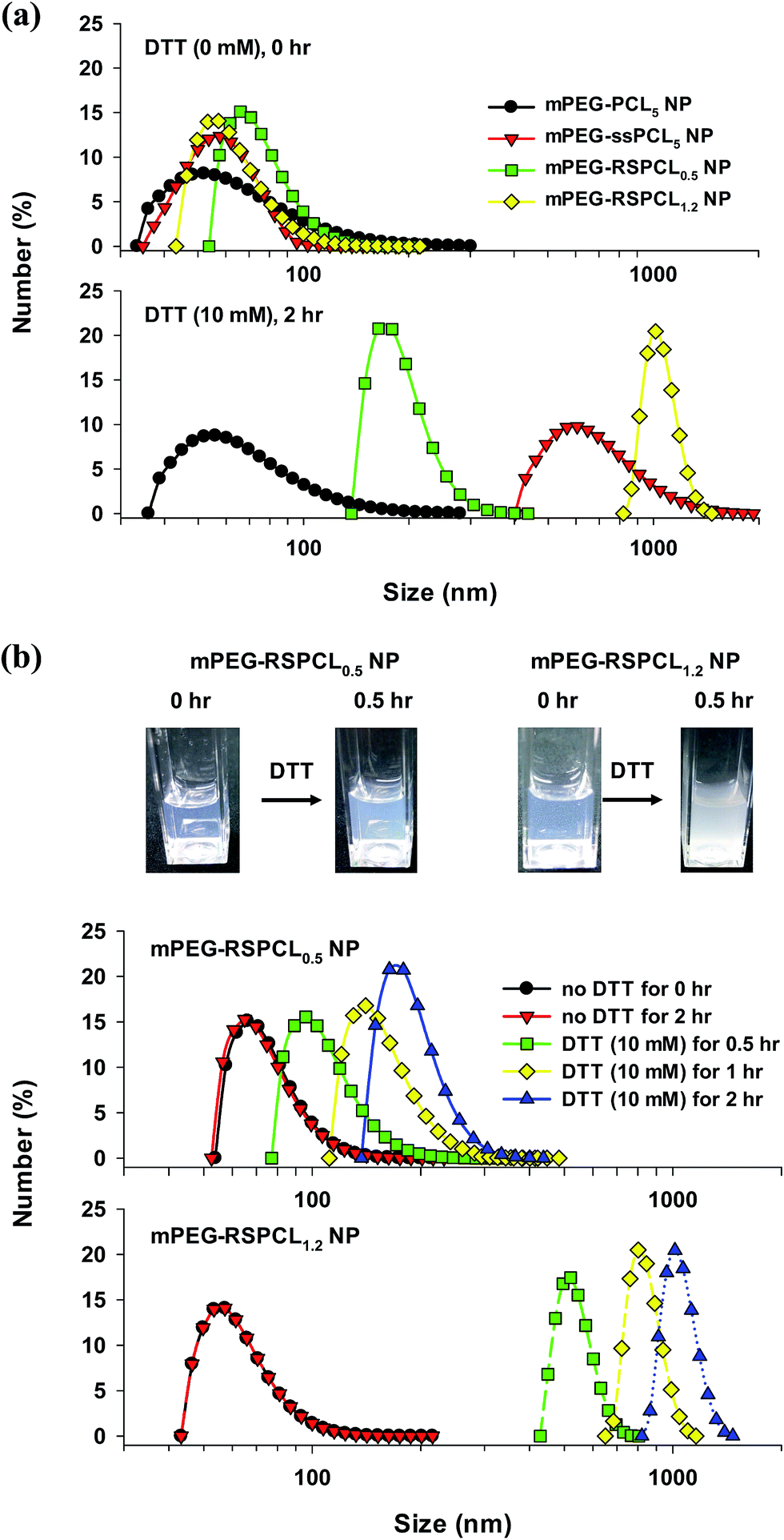 | ||
| Fig. 5 (a) Thiol-induced size change of various NPs and (b) time-dependent size change of mPEG-RSPCL0.5 NPs and mPEG-RSPCL1.2 NPs in DTT-containing DPBS (50 mM, pH 7.4). The degradation experiment was performed at 37 °C. ([polymer] = 0.25 mg mL−1, [DTT] = 10 mM). | ||
In general, when developing nanosized drug delivery carriers, good biocompatibility and negligible cytotoxicity are required. Although many studies have reported negligible cytotoxicity of PCL-based drug delivery systems,11–13,33 the cytotoxicity of mPEG-RSPCL NPs in HeLa and HepG2 cell lines was examined. The mPEG-PCL5 NPs and mPEG-ssPCL5 NPs as the control NPs represented above 90% cell viability in all concentrations of the NPs tested in this study (i.e., 6.25–400 μg mL−1) (Fig. S6†). In HeLa cells, 80% cell viability was observed in the presence of mPEG-RSPCL0.5 and mPEG-RSPCL1.2 NPs below 50 μg mL−1, but cell viabilities approached 60% at 400 μg mL−1 (Fig. 6). mPEG-RSPCL1.2 NP-treated HepG2 cells had greater than 80% cell viability in 6.25–400 μg mL−1. However, HepG2 cell viability in <100 μg mL−1 of mPEG-RSPCL0.5 NPs was greater than 80%, but was approximately 60% at 400 μg mL−1, which is similar to mPEG-RSPCL0.5 and mPEG-RSPCL1.2 NPs at the same concentration in HeLa cells. Thus, the cytotoxicity of mPEG-RSPCL NPs may not be significantly affected by the MWs of the RSPCL polymers and the PCL diol; additionally, the mPEG-RSPCL NPs showed negligible cytotoxicity below 200 μg mL−1.
 | ||
| Fig. 6 In vitro cytotoxicity of drug-free mPEG-RSPCL NPs in (a) HeLa and (b) HepG2 cells at 48 h post-treatment. Various concentrations of NPs were used to treat the cells for 48 h, and the data are expressed as the mean ± standard error of the mean (SEM) (n = 12). | ||
Preparation and physicochemical, morphological, and drug-releasing characteristics of drug-loaded mPEG-RSPCL NPs
It was expected that mPEG-RSPCL NPs may act as delivery carriers of therapeutics because mPEG-RSPCL NPs showed negligible cytotoxicity under a reasonable concentration range for their applications (Fig. 6) and were formed by a self-assembly (Table 2). Thus, the drug encapsulation capability of mPEG-RSPCL NPs was evaluated with loading of either a hydrophilic or a hydrophobic drug. Additionally, the types of drug-loaded nanostructures formed depending on the physical characteristics of the loadable payload were predicted. To pursue the physical loading of two different hydrophilic or hydrophobic therapeutics, a co-solvent dispersion (CD) was used for loading of the hydrophobic drug during the preparation of mPEG-RSPCL NPs, whereas a remote loading technique using an ammonium sulfate (AS) gradient was applied to encapsulate a hydrophilic drug after constructing the mPEG-RSPCL NPs. To compare the therapeutic efficacy of the hydrophilic and hydrophobic drugs, hydrophobic salt-free DOX and its hydrochloride salt-form (i.e., DOX·HCl) were selected as model drugs.First, a hydrophobic DOX was loaded into mPEG-RSPCL NPs forming DOX-mPEG-RSPCL0.5 and DOX-mPEG-RSPCL1.2 NPs. Their sizes and zeta-potentials were approximately 80–90 nm in diameter and nearly neutral, respectively (Table 2), and were not significantly different from those of the drug-free mPEG-RSPCL NPs. When the drug loading content (DLC) of DOX in mPEG-RSPCL NPs was targeted at 10 wt%, the actual DLC and drug loading efficiency (DLE) were approximately 5.2–5.4 wt% and 52–54%, respectively (Table 2).
Second, a hydrophilic DOX·HCl was loaded into the formed mPEG-RSPCL NPs via an AS gradient. After removing the unloaded DOX·HCl, the measured sizes of the resulting NPs were approximately 30–35 nm larger than drug-free NPs, and the drug loading process did not make a remarkable difference in the zeta-potential (Table 2). The size and zeta-potential results did not confirm that the resulting NPs contained a hydrophilic DOX·HCl. However, further evaluation of the DLCs and DLEs supported the formation of drug-loaded NPs. That is, the DLC and DLE of DOX·HCl-mPEG-RSPCL0.5 NPs were approximately 4.5 wt% and 45%, respectively, and the DLC and DLE of DOX·HCl-mPEG-RSPCL1.2 NPs were approximately 4.2 wt% and 42%, respectively (Table 2). Although DOX·HCl-loaded NPs had somewhat lower DLC and DLE than DOX-loaded NPs, the results indicate successful drug loading of DOX·HCl in mPEG-RSPCL NPs.
Based on the successful loading of water-insoluble DOX in mPEG-RSPCL NPs, it was expected that a hydrophobic DOX could be placed in either a hydrophobic core of micellar nanostructures or a hydrophobic space in a bilayer of nanosized vesicular structures. Additionally, it was thought that mPEG-RSPCL NPs could be polymeric vesicles because the AS gradient method allowed successful DOX·HCl loading in mPEG-RSPCL NPs. Thus, to confirm the nanostructures of the drug-loaded mPEG-RSPCL NPs, their morphologies were evaluated by cryoTEM. Interestingly, unlike our expectation of polymeric vesicles, drug-free mPEG-RSPCL NPs, DOX·HCl-mPEG-RSPCL NPs, and DOX-mPEG-RSPCL NPs had no layers for polymersomes (Fig. 7 and S7†). That is, all NPs were micellar structures, regardless of the NP preparation and the drug hydrophilicity/hydrophobicity. The estimated volume fraction of mPEG (fmPEG) values for mPEG-RSPCL0.5 and mPEG-RSPCL1.2 copolymers were approximately 0.7 and 0.56, respectively, based on the densities of the bulk polymers (ρPEG = 1.13 g cm−3, ρPCL = 1.25 g cm−3),13 and this supported spherical micelle structures of mPEG-RSPCL0.5 and mPEG-RSPCL1.2 NPs based on literature results.39
 | ||
| Fig. 7 CryoTEM images of drug-free mPEG-RSPCL0.5 and drug-loaded mPEG-RSPCL0.5 NPs. | ||
Although micellar nanostructures of mPEG-RSPCL NPs were confirmed by cryoTEM images, it was still questioned how hydrophilic DOX·HCl was loaded into the NPs. The explanation may be due to a higher temperature (around melting temperature (Tm))-induced water channel formation in NPs. DLC and DLE were increased as the loading temperature was increased (Fig. S8†). A 60 °C condition was used when applying the AS gradient, which was higher than Tm (around 55 °C) of PCL. In general, a temperature above Tm causes increased free volume and water channels in hydrophobic matrix, resulting in faster release of the loaded hydrophilic drugs from the matrix. Similarly, it is expected that the temperature (60 °C) used during the loading of DOX·HCl might make more and larger water channels in the hydrophobic and induced-anionic RSPCL core in mPEG-RSPCL NPs, and the hydrophilic and cationic DOX·HCl molecules might be spontaneously moved through the aqueous channels and into the NPs. After loading DOX·HCl into the NPs, RT or 4 °C conditions might close or reduce the free volume and water channels in the RSPCL core of mPEG-RSPCL NPs. Thus, it is suggested that DOX-mPEG-RSPCL NPs had a somewhat higher DLC than DOX·HCl-mPEG-RSPCL NPs and that the loading of DOX·HCl resulted in approximately 30–35 nm larger NPs compared to the size of drug-free NPs because the dispersion of hydrophilic DOX·HCl molecules in the hydrophobic RSPCL core could interfere with the hydrophobic interaction among RSPCL chains in the core.
In addition, another possible explanation for the loading of the hydrophilic drug (i.e., DOX·HCl) into the micelle is the formation of large complex micelles (LCMs). In general, LCMs have larger sizes than true micelles with hydrophilic corona and one hydrophobic core and also have hydrophilic channels or domains in their core2 because DOX·HCl-loaded micelles are relatively larger than drug-free micelles and inverted micellar structures in LCMs exist. Thus, hydrophilic DOX·HCl might be loaded into hydrophilic domains in LCMs.
Thiol-triggered release behavior of drug-loaded mPEG-RSPCL NPs
To measure drug release from drug-loaded mPEG-RSPCL NPs in dialysis device at 37 °C, DOX·HCl as a model drug were used. In addition, to compare the effects of drug release on the presence and the number of disulfide bonds in NPs, three NPs (i.e., mPEG-PCL5 NPs, mPEG-ssPCL5 NPs, and mPEG-RSPCL0.5 NPs) were prepared from their corresponding polymers having similar MWs. About 70% of free DOX·HCl was permeated within 3 h and almost all of free DOX·HCl was dialyzed out within 10 h. However, DOX·HCl-loaded NPs showed different release patterns depending on the presence of DTT. As shown in Fig. 8(a) and (b), the drug release behavior of mPEG-PCL5 NPs was not affected by the presence of DTT because the NPs did not have a disulfide bond. In the absence of DTT, mPEG-ssPCL5 NPs and mPEG-RSPCL0.5 NPs having disulfide bonds released below 10% of the initial drug for 12 h and their drug release patterns are almost identical to that of mPEG-PCL5 NPs (Fig. 8(a)), suggesting that the presence and the number of the disulfide bonds in NPs did not affect drug release profiles. However, the presence of thiols (i.e., 10 mM of DTT) caused different drug release rates from three NPs: mPEG-RSPCL0.5 NPs > mPEG-ssPCL5 NPs > mPEG-PCL5 NPs in the fastest-to-slowest order (Fig. 8(b)). Thiol-triggered drug release rates could be related with size of hydrophobic fragments after thiol-triggered cleaving hydrophobic PCL cores in NPs because breaking more disulfide bonds in NPs could form smaller hydrophobic fragments. | ||
| Fig. 8 Time-dependent thiol-triggered drug release of DOX·HCl-loaded NPs in DIW at 37 °C. The data were expressed as the mean ± standard deviation (n = 3). | ||
In vitro anti-tumor characteristics of drug-loaded mPEG-RSPCL NPs
The mPEG-RSPCL NPs encapsulated both a hydrophobic DOX and a hydrophilic DOX·HCl (Table 2). Thus, to examine whether mPEG-RSPCL NPs efficiently deliver DOX and its salt form to cells, the therapeutic efficiency of drug-loaded mPEG-RSPCL NPs was evaluated by the MTT-based cytotoxicity assay in HeLa and HepG2 cells. In HeLa cells, DOX·HCl-mPEG-RSPCL0.5 NPs and DOX·HCl-mPEG-RSPCL1.2 NPs killed half of the cells at approximately 0.47 and 0.26 μg mL−1, respectively, and their IC50 (the drug concentration that causes 50% growth inhibition) values were approximately 2- and 3.6-fold lower than that of free DOX·HCl (IC50 ≈ 0.93 μg mL−1) (Fig. 9(a)). DOX·HCl-mPEG-RSPCL0.5 NPs showed superior killing effects to two control DOX·HCl-mPEG-ssPCL5 NPs (IC50 ≈ 0.75 μg mL−1) and DOX·HCl-mPEG-PCL5 NPs (IC50 ≈ 2 μg mL−1). The results indicate that thiol-triggered DOX·HCl releasing NPs represented more potent anti-tumor effects than free DOX·HCl unlike thiol-insensitive DOX·HCl releasing NPs. Similarly, in HepG2 cells, DOX·HCl-mPEG-RSPCL0.5 NPs, DOX·HCl-mPEG-RSPCL1.2 NPs, and DOX·HCl-mPEG-ssPCL5 NPs had approximately 1.7-fold, 2-fold, and 1.3-fold better anti-tumor activity than free DOX·HCl (IC50 ≈ 0.58 μg mL−1), whereas DOX·HCl-mPEG-PCL5 NPs had IC50 ≈ 1.8 μg mL−1 (Fig. 9(b)).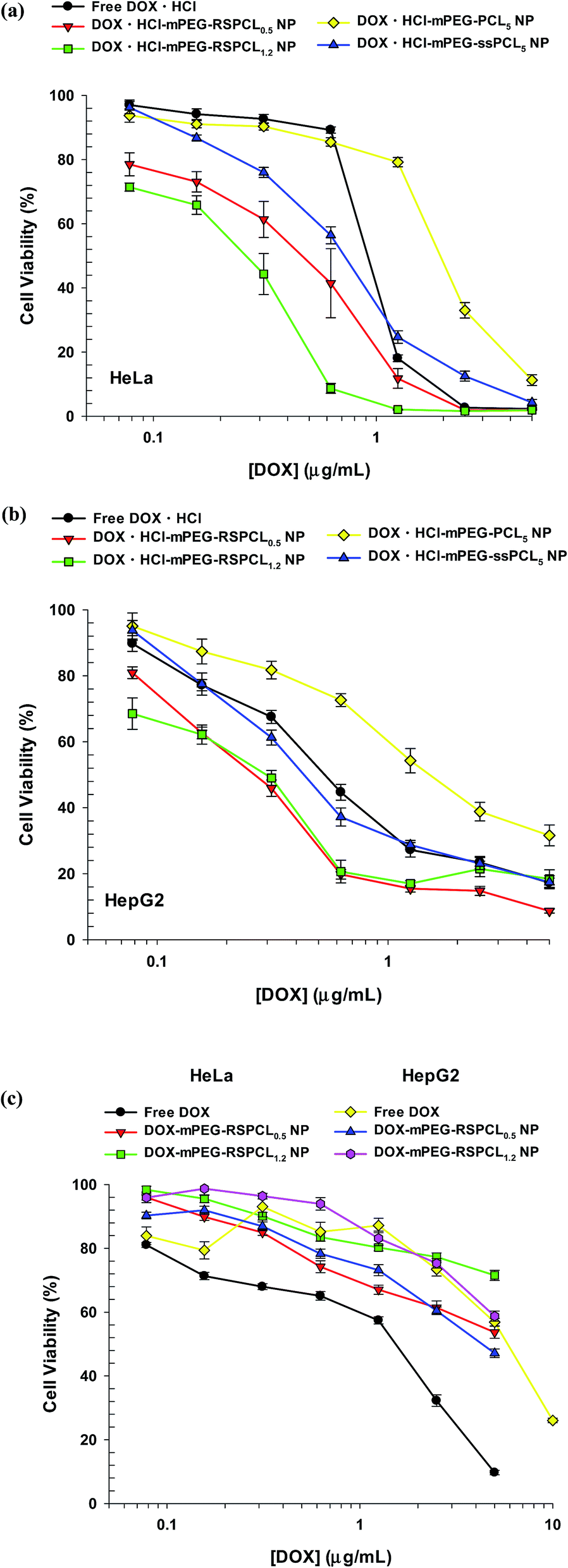 | ||
| Fig. 9 In vitro anti-tumor effects of DOX·HCl-loaded mPEG-RSPCL NPs in (a) HeLa and (b) HepG2 cells and DOX-loaded mPEG-RSPCL NPs in (c) HeLa and HepG2 cells at 48 h post-treatment. Various concentrations of drug-loaded NPs and free drugs were used to treat the cells for 48 h, and the data are expressed as the mean ± SEM (n = 12–24). | ||
In the case of DOX-loaded NPs, the IC50 value of free DOX was 1.54 μg mL−1, whereas approximately 71 and 54% of DOX-mPEG-RSPCL0.5 NP-treated and DOX-mPEG-RSPCL1.2 NP-treated HeLa cells, respectively, were viable at the highest DOX concentration (5 μg mL−1) (Fig. 9(c)). In HepG2 cells, DOX-mPEG-RSPCL0.5 and DOX-mPEG-RSPCL1.2 NPs killed approximately 58 and 47% of the cells, respectively, at the highest DOX concentration (5 μg mL−1) in this study, and their dose-dependent cytotoxicity was similar to that of free DOX (IC50 ≈ 5.8 μg mL−1) (Fig. 9(c)).
Although it is unclear that these anti-tumor results were caused by different aqueous solubilities between DOX and DOX·HCl40 (1.18 mg mL−1 vs. 10 mg mL−1 reported in http://www.drugbank.ca/drugs) and/or an inactive dimeric form of DOX,41 free DOX·HCl is more potent than free DOX. Although DOX-mPEG-RSPCL NPs had similar or inferior anti-tumor effects to free DOX, DOX·HCl-mPEG-RSPCL NPs had much better drug effects than free DOX·HCl. Particularly, DOX·HCl-loaded NPs had much higher cytotoxic activity than DOX-loaded NPs. That is, DOX·HCl-mPEG-RSPCL0.5 and DOX·HCl-mPEG-RSPCL1.2 NPs showed approximately 11- to 13-fold and at least 17-fold, respectively, superior anti-tumor efficacy compared with their DOX-loaded counterpart NPs (Fig. 9).
Cellular uptake and intracellular distribution of drug-loaded mPEG-RSPCL NPs
Although DOX·HCl-mPEG-RSPCL and DOX-mPEG-RSPCL NPs were prepared from the same mPEG-RSPCL copolymers, their drug efficacies were completely different (Fig. 9). Thus, their cellular uptakes were assessed because drug- or NP-mediated anti-tumor effects are strongly affected by cellular uptake. To quantify the amount of intracellular DOX, flow cytometry was used after treating either free drugs (i.e., DOX and DOX·HCl) or drug-loaded mPEG-RSPCL NPs at [DOX] = 5 μg mL−1 in HeLa and HepG2 cells. In this study, it was expected that hydrophobic DOX would be taken up more rapidly through the plasma membrane via adsorptive endocytosis than the relatively hydrophilic DOX·HCl because hydrophobic molecules interact more with the plasma membrane than hydrophilic molecules. However, interestingly, at 4 h post-treatment, free DOX·HCl showed approximately 3- and 1.7-fold more cellular uptake (based on fluorescence intensity) than free DOX in HeLa and HepG2 cells, respectively (Fig. 10). As previously reported,40 it is likely explained by different aqueous solubilities of free drugs: DOX·HCl is water-soluble, while DOX is water-insoluble. In addition, it could be described by an inactive DOX dimer because the fluorescence intensity of DOX dimer is much lower than that of DOX monomer.42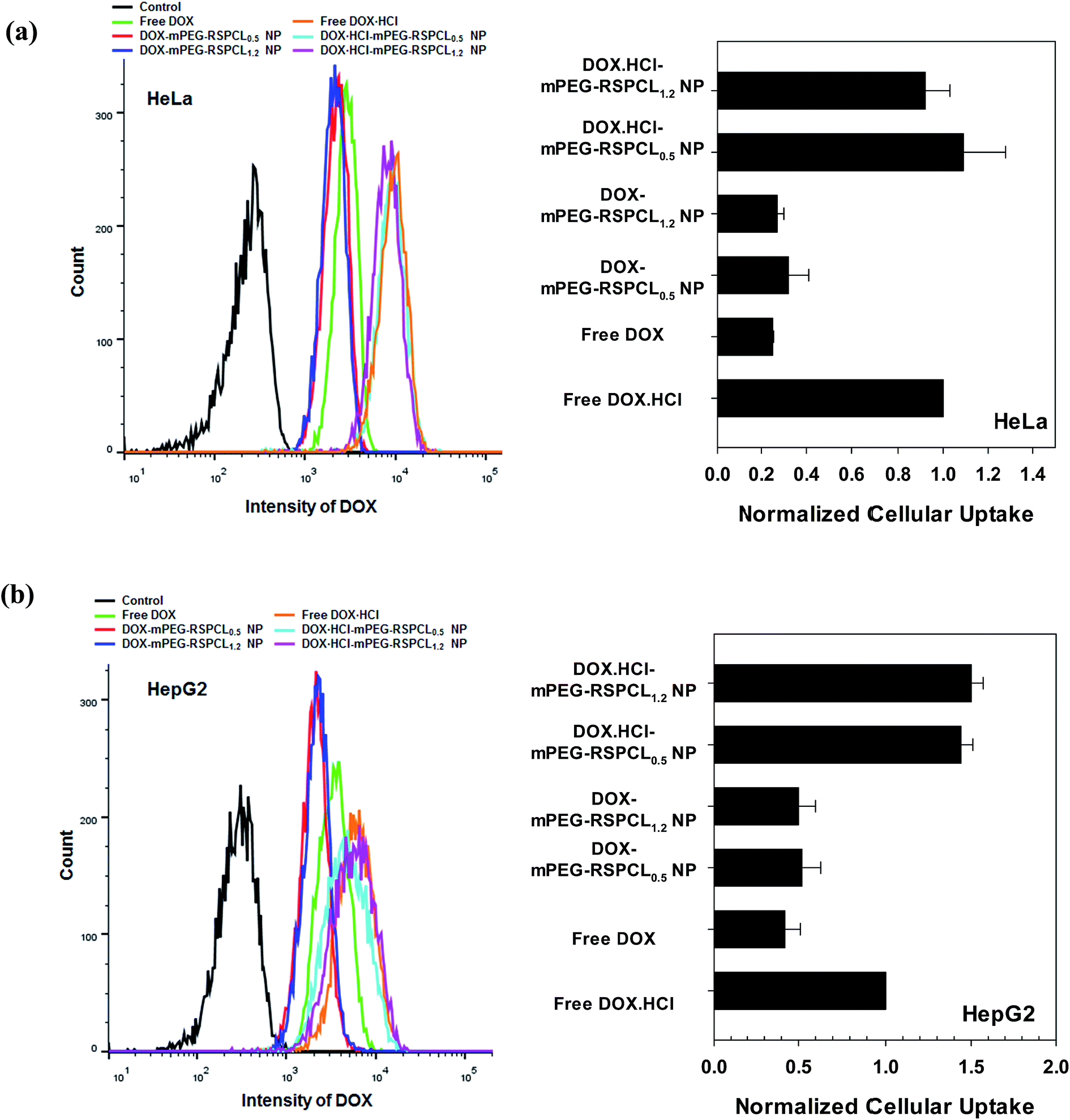 | ||
| Fig. 10 Cellular uptake of drug-loaded mPEG-RSPCL NPs in (a) HeLa and (b) HepG2 cells at 4 h post-treatment. The cells were treated with drug-loaded NPs and free drugs ([drug] = 5 μg mL−1) for 4 h. | ||
It was expected that the cellular uptake of drug-loaded PEGylated mPEG-RSPCL NPs would be lower than the free drugs because it was known that PEG-decorated NPs interfere with cellular uptake and also that PEGylated NPs without cell targeting moieties typically follow slow fluid-phase endocytosis.43,44 Contrary to our expectation, DOX·HCl-mPEG-RSPCL NPs showed similar or slightly superior (∼1.5-fold) cellular uptake than that of free DOX·HCl in HeLa and HepG cells (Fig. 10). Cellular uptake of DOX-mPEG-RSPCL NPs was similar to that of free DOX in both cell lines.
In addition, it was expected that DOX·HCl-mPEG-RSPCL NPs would have similar cellular uptake as DOX-mPEG-RSPCL NPs because their cellular uptakes may be dependent upon the carrier but not the drug. However, the cellular uptakes of DOX·HCl-mPEG-RSPCL NPs were not same to those of DOX-mPEG-RSPCL NPs in both cell lines. Namely, the cellular uptake of DOX·HCl-mPEG-RSPCL NPs was on average 3.4- and 2.9-fold greater than that of DOX-mPEG-RSPCL NPs in HeLa and HepG2 cells, respectively (Fig. 10). The results may be due to different aqueous solubilities of free drugs, as mentioned above. Although similar amounts of the free drugs were delivered with the NPs, their different aqueous solubilities may further affect molecular aggregation of the relatively hydrophobic drugs resulting in lower fluorescence.42
In HeLa cells, the cellular uptakes of drug-loaded mPEG-RSPCL NPs were further examined by confocal microscopy (Fig. 11). Similar to the previous results (Fig. 10(a)), DOX·HCl-mPEG-RSPCL NPs and free DOX·HCl had similar fluorescence in the cells and had stronger fluorescence than DOX-mPEG-RSPCL NPs. In particular, DOX·HCl delivered with or without mPEG-RSPCL NPs was largely located in the nuclear region.
 | ||
| Fig. 11 Intracellular distribution of drug-loaded mPEG-RSPCL NPs in HeLa cells at 4 h post-treatment. The cells were treated with drug-loaded NPs and free drug ([drug] = 5 μg mL−1) for 4 h. | ||
Although the use of hydrophobic DOX and hydrophilic DOX·HCl has been frequently reported, it was difficult to find the comparison of anti-tumor effects of hydrophobic DOX and hydrophilic DOX·HCl. Xu et al., reported that DOX·HCl-loaded NPs had better anti-tumor effects than DOX-loaded NPs, however the explanation of this result was unclear.45 In this study, we observed different efficacies of these two drugs because mPEG-RSPCL NPs were able to successfully load both chemical drugs. Regardless of the use of drug carriers, the different drug efficacies between DOX and DOX·HCl may be influenced by cellular uptake and intracellular distribution because the lower cellular uptake of free DOX resulted in lower anti-tumor activity than free DOX·HCl. Similarly, the lower cellular uptake of DOX-mPEG-RSPCL NPs resulted in lower anti-tumor activity than DOX·HCl-mPEG-RSPCL NPs. Additionally, the resulting cellular uptake of DOX·HCl-mPEG-RSPCL NPs may have higher anti-tumor effects than free DOX·HCl. Although many researchers have used hydrophobic DOX for drug loading in micelles, our results suggest that hydrophilic DOX·HCl may be more potent and efficacious than hydrophobic DOX in nanocarriers.
Conclusion
Thiol-triggered degradable mPEG-RSPCL copolymers with multiple disulfide bonds were synthesized and formed self-assembled NPs in aqueous environments. The resulting mPEG-RSPCL NPs with negligible cytotoxicity were degraded and changed in size by thiol treatment. The mPEG-RSPCL NPs loaded either a hydrophobic DOX or its hydrophilic DOX·HCl, resulting in drug-loaded mPEG-RSPCL NPs with an approximate 100 nm diameter and an almost neutral surface. Depending on the loaded drugs, DOX·HCl-mPEG-RSPCL NPs showed superior anti-tumor effects to free DOX·HCl, whereas DOX-mPEG-RSPCL NPs were similar or inferior therapeutically to free DOX. The drug efficacy of the drug-loaded mPEG-RSPCL NPs is strongly influenced by cellular uptake. In conclusion, the mPEG-RSPCL NPs, which are able to encapsulate two different hydrophilic and hydrophobic drugs, show potential for the intracellular delivery of therapeutic agents.Experimental
Materials
Polycaprolactone diols having MW 1250 Da (PCL1.2 diol), MW 530 Da (PCL0.5 diol), and MW 5000 Da (PCL5 diol) were purchased from Polysciences, Inc. (Warrington, PA, USA), Sigma-Aldrich Company (St. Louis, MO, USA), and CM-Tec, Inc. (Dewark, DE, USA), respectively. Polycaprolactone dicarboxylic acid having MW 5000 Da (PCL5 COOH) was bought from CM-Tec, Inc. (Dewark, DE, USA). Poly(ethylene glycol) methyl ether (mPEG, Mn 5000 Da), 3,3′-dithiodipropionic acid (DTPA), N,N′-dicyclohexylcarbodiimide (DCC), 4-(dimethylamino)pyridine (DMAP), triethylamine (TEA), chloroform, ammonium sulfate (AS), DL-dithiothreitol (DTT), RPMI-1640 medium, Dulbecco's Modified Eagle's Medium (DMEM), sodium bicarbonate, D-glucose, Ca2+-free and Mg2+-free Dulbecco's phosphate buffered saline (DPBS), 4-(2-hydroxy-ethyl)-1-piperazine (HEPES), fetal bovine serum (FBS), penicillin–streptomycin antibiotics, trypsin–EDTA, dimethyl sulfoxide (DMSO), anhydrous tetrahydrofuran (THF), dichloromethane (DCM), hexane, diethyl ether, methanol, Hoechst33342, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich Company (St. Louis, MO, USA). Doxorubicin hydrochloride (DOX·HCl) was thankfully donated by the Ilyang Pharmaceutical Company (Seoul, Republic of Korea) or purchased from MedKoo Biosciences (Chapel Hill, NC, USA).Synthesis and chemical characterization of HMW RSPCL polymers
To obtain HMW RSPCL, LMW PCL diols (PCL1.2 diol and PCL0.5 diol) and the single disulfide-containing DTPA were alternatively connected by a conjugation reaction between two hydroxyl groups of LMW PCL diol blocks and two carboxylic acids of DTPA molecules (Fig. 2(a)). In brief, a solution of DCC (3.6 mmol) and DMAP (3.6 mmol) dissolved in THF (5 mL) were slowly dropped into a solution of LMW PCL diol (1 mmol) and DTPA (1.5 mmol) dissolved in THF (10 mL). In the presence of TEA (0.2 mL) as a catalyst, the reaction was performed at room temperature (RT) under N2 gas for 2 d. The polymer solution with the synthesized RSPCL was filtered to remove dicyclohexylurea (DCU) and the solvent (THF) was evaporated. The dried polymers were dissolved in DCM and were filtered to completely remove the remaining traces of DCU. The clear filtrate was precipitated in hexane twice and the resulting precipitate (RSPCL polymer) was dried in vacuo (sticky yellowish paste and 74.5% for RSPCL0.5; white solid and 75.4% for RSPCL1.2).After dissolving the synthesized RSPCL polymers in CDCl3, their chemical structures and compositions were confirmed using a 500 MHz Bruker 1H-NMR spectrometer (Billerica, MA, USA). Detailed peaks of RSPCL polymers in 1H-NMR spectra were assigned as follows and most protons in their polymers were represented as broad and multiplet peaks: for RSPCL polymers (CDCl3, δ, ppm), 3.7 (–C(O)OCH2CH2OCH2CH2OC(O)– of PCL, a), 4.25 (C(O)OCH2CH2OCH2CH2OC(O)– of PCL, b), 2.33 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, c), 1.6–1.7 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, d and f), 1.38 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, e), 4.08 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, g), 2.74 (–OC(O)CH2CH2SSCH2CH2C(O)O– of DTPA, h), and 2.92 (–OC(O)CH2CH2SSCH2CH2C(O)O– of DTPA, i).
Synthesis and chemical characterization of mPEG-RSPCL copolymers
Two carboxylic acids at two ends of the synthesized RSPCL polymers, which represent a DTPA-(PCL-DTPA)x structure, were reacted with one hydroxyl group at one end of mPEG by a coupling reaction and the resulting mPEG-RSPCL copolymers were a triblock-type structure (i.e., mPEG-b-RSPCL-b-mPEG copolymer) (Fig. 2(a)). In brief, RSPCL polymers (0.05 mmol) and mPEG (0.12 mmol) were dissolved in chloroform (15 mL). A solution of DCC (0.144 mmol) and DMAP (0.144 mmol) dissolved in chloroform (5 mL) was added into the polymer solution. In the presence of TEA (40 μL), the coupling reaction between mPEG and RSPCL was carried out at RT for 2 d. The copolymer solution was filtered to remove DCU and the filtrate was slowly dropped into cold diethyl ether to precipitate the synthesized mPEG-RSPCL copolymers. Then, the precipitate was filtered, washed with methanol to remove any unreacted mPEG, and then dried in vacuo. The resulting mPEG-RSPCL copolymers dissolved in DMSO were dialyzed against deionized water (DIW) using a dialysis membrane (molecular weight cut-off (MWCO) 8 kDa) for 2 d. The formed mPEG-RSPCL copolymers were filtered with a 0.45 μm syringe filter and then were lyophilized (white solid and 51.6% for mPEG-RSPCL0.5; white solid and 54.9% for mPEG-RSPCL1.2). Broad and multiplet peaks of mPEG-RSPCL copolymers (CDCl3, δ, ppm) in 1H-NMR spectra were assigned as follows: 3.69 (–C(O)OCH2CH2OCH2CH2OC(O)– of PCL, a), 4.19 (C(O)OCH2CH2OCH2CH2OC(O)– of PCL, b), 2.32 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, c), 1.58–1.7 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, d and f), 1.38 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, e), 4.06 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, g), 2.73 (–OC(O)CH2CH2SSCH2CH2C(O)O– of DTPA, h), 2.92 (–OC(O)CH2CH2SSCH2CH2C(O)O– of DTPA, i), 4.19 (–OCH2CH2OC(O)– of mPEG, j), 3.69 (–OCH2CH2OC(O)– of mPEG, k), 3.64 (–OCH2CH2O– of mPEG, l and m), and 3.38 (–OCH3 of mPEG, n).
Synthesis and chemical characterization of mPEG-ssPCL5 copolymers and mPEG-PCL5 copolymers
To obtain HOOC-ssPCL5-ss-COOH (ssPCL5ss), one PCL5 diol was reacted with excess of the single disulfide-containing DTPA (Fig. 2(b)). In brief, a solution of DCC (1 mmol) and DMAP (1 mmol) dissolved in THF (5 mL) were slowly dropped into a solution of PCL5 diol (0.2 mmol) and DTPA (0.5 mmol) dissolved in THF (10 mL). In the presence of TEA (0.05 mL) as a catalyst, the reaction was performed at room temperature (RT) under N2 gas for 2 d. The polymer solution with the synthesized ssPCL5ss was filtered to remove DCU and the solvent (THF) was evaporated. The dried polymers were dissolved in DCM and were filtered to completely remove the remaining traces of DCU. The clear filtrate was precipitated in hexane twice and the resulting precipitate (ssPCL5ss polymer) was dried in vacuo (white solid and 88.6%). Broad and multiplet peaks of ssPCL5ss polymer (CDCl3, δ, ppm) in 1H-NMR spectra were assigned as follows: 3.73 (–C(O)OCH2CH2OCH2CH2OC(O)– of PCL, a), 4.27 (C(O)OCH2CH2OCH2CH2OC(O)– of PCL, b), 2.34 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, c), 1.62–1.74 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, d and f), 1.42 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, e), 4.08 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, g), 2.76 (HOC(O)CH2CH2SSCH2CH2C(O)O– of DTPA, h), and 2.92 (HOC(O)CH2CH2SSCH2CH2C(O)O– of DTPA, i).
Two carboxylic acids at two ends of the synthesized ssPCL5ss polymers were reacted with one hydroxyl group at one end of mPEG by a coupling reaction and the resulting mPEG-ssPCL5 copolymers were a triblock-type structure (i.e., mPEG-b-ssPCL5ss-b-mPEG copolymer) (Fig. 2(b)). In brief, ssPCL5ss polymers (0.05 mmol) and mPEG (0.12 mmol) were dissolved in chloroform (15 mL). A solution of DCC (0.144 mmol) and DMAP (0.144 mmol) dissolved in chloroform (5 mL) was added into the polymer solution. In the presence of TEA (40 μL), the coupling reaction between mPEG and ssPCL5ss was carried out at RT for 2 d. The copolymer solution was filtered to remove DCU and the filtrate was slowly dropped into cold diethyl ether to precipitate the synthesized mPEG-ssPCL5 copolymers. Then, the precipitate was filtered, washed with methanol to remove any unreacted mPEG, and then dried in vacuo. The resulting mPEG-ssPCL5 copolymers dissolved in DMSO were dialyzed against DIW using a dialysis membrane (MWCO 8 kDa) for 2 d. The formed mPEG-ssPCL5 copolymers were filtered with a 0.45 μm syringe filter and then were lyophilized (white solid and 67.4%). Broad and multiplet peaks of mPEG-ssPCL5 copolymers (CDCl3, δ, ppm) in 1H-NMR spectra were assigned as follows: 3.68 (–C(O)OCH2CH2OCH2CH2OC(O)– of PCL, a), 4.23 (C(O)OCH2CH2OCH2CH2OC(O)– of PCL, b), 2.31 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, c), 1.6–1.69 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, d and f), 1.39 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, e), 4.06 (–C(O)OCH2CH2CH2 CH2CH2C(O)O– of PCL, g), 2.74 (–OC(O)CH2CH2SSCH2CH2C(O)O– of DTPA, h), 2.91 (–OC(O)CH2CH2SSCH2CH2C(O)O– of DTPA, i), 4.23 (–OCH2CH2OC(O)– of mPEG, j), 3.69 (–OCH2CH2OC(O)– of mPEG, k), 3.64 (–OCH2CH2O– of mPEG, l and m), and 3.38 (–OCH3 of mPEG, n).
For synthesis of mPEG-PCL5 copolymers, two carboxylic acids in the commercially-available HOOC-PCL5-COOH were reacted with one hydroxyl group at one end of mPEG by a coupling reaction and the resulting mPEG-PCL5 copolymers were a triblock-type structure (i.e., mPEG-b-PCL5-b-mPEG copolymer) (Fig. 2(b)). In brief, HOOC-PCL5-COOH (0.05 mmol) and mPEG (0.12 mmol) were dissolved in chloroform (15 mL). A solution of DCC (0.144 mmol) and DMAP (0.144 mmol) dissolved in chloroform (5 mL) was added into the polymer solution. In the presence of TEA (40 μL), the coupling reaction between mPEG and HOOC-PCL5-COOH was carried out at RT for 2 d. The copolymer solution was filtered to remove DCU and the filtrate was slowly dropped into cold diethyl ether to precipitate the synthesized mPEG-PCL5 copolymers. Then, the precipitate was filtered, washed with methanol to remove any unreacted mPEG, and then dried in vacuo. The resulting mPEG-PCL5 copolymers dissolved in DMSO were dialyzed against DIW using a dialysis membrane (MWCO 8 kDa) for 2 d. The formed mPEG-PCL5 copolymers were filtered with a 0.45 μm syringe filter and then were lyophilized (white solid and 81.3%). Broad and multiplet peaks of mPEG-PCL5 copolymers (CDCl3, δ, ppm) in 1H-NMR spectra were assigned as follows: 3.68 (–C(O)OCH2CH2OCH2CH2OC(O)– of PCL, a), 4.23 (C(O)OCH2CH2OCH2CH2OC(O)– of PCL, b), 2.31 (–C(O)OCH2CH2CH2 CH2CH2C(O)O– of PCL, c), 1.61–1.69 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, d and f), 1.38 (–C(O)OCH2CH2CH2CH2CH2C(O)O– of PCL, e), 4.06 (–C(O)OCH2CH2CH2 CH2CH2C(O)O– of PCL, g), 4.23 (–OCH2CH2OC(O)– of mPEG, h), 3.68 (–OCH2CH2OC(O)– of mPEG, i), 3.64 (–OCH2CH2O– of mPEG, j and k), and 3.38 (–OCH3 of mPEG, l).
Physical characterization of RSPCL polymers and mPEG-RSPCL copolymers
To evaluate the molecular weights (MWs), molecular weight distributions (MWDs), and polydispersity indices (PDIs) of the synthesized RSPCL polymers and mPEG-RSPCL copolymers, the synthesized polymers were dissolved in THF and were analyzed by high-performance liquid chromatography (HPLC) system (Agilent 1260 infinity series) equipped with a gel permeation chromatography (GPC) analysis mode (Agilent Technologies, Santa Clara, CA, USA). For GPC analysis, a PLgel column (5 μm Mixed-D 300 × 7.5 column; Agilent Technologies, Santa Clara, CA, USA) was used and THF was eluted at a flow rate of 1.0 mL min−1 at 35 °C. MWs, MWDs, and PDIs of the synthesized RSPCL polymers and mPEG-RSPCL copolymers were analyzed by comparison with a MW standard curve of monodisperse polystyrenes. For the control copolymers (i.e., mPEG-PCL5 and mPEG-ssPCL5), their MW characteristics were similarly evaluated. Detailed GPC data of RSPCL polymers, mPEG-RSPCL copolymers, ssPCL5ss polymer, mPEG-ssPCL5 copolymer, and mPEG-PCL5 copolymer were summarized in Table 1.Preparation and physicochemical and degradation characterization of drug-free mPEG-RSPCL nanoparticles (NPs)
The mPEG-RSPCL copolymers (4 mg) were dissolved in DMSO (0.2 mL) and then DIW (3.8 mL) was quickly added into the polymer solution under vigorous stirring. The mPEG-RSPCL copolymers were self-assembled to construct their nanostructures and then the formed nanoparticles (NPs) were further dialyzed against DIW using a dialysis membrane (MWCO 2 kDa) for 24 h to remove DMSO and were then filtered with a 0.45 μm syringe filter. The particle size and zeta-potential of the resulting mPEG-RSPCL NPs (0.25 mg mL−1 diluted from 1 mg mL−1) were monitored by a zeta-potential and particle size analyzer (ELS-Z; Photal Otsuka Electronics Co., Osaka, Japan) with a fixed wavelength of 677 nm and a constant angle of 90° at RT. For comparison, the particle size and zeta-potential of the control NPs were similarly evaluated.To evaluate the reduction-sensitive size change of drug-free mPEG-RSPCL NPs, the NPs (0.25 mg mL−1) were exposed to DTT (1 μM to 100 mM) in DPBS (1 mL, 50 mM, pH 7.4). At pre-determined time points, their time-dependent size changes were detected by a particle size analyzer (ELS-Z; Photal Otsuka Electronics Co., Osaka, Japan). For comparison, the sizes of the control NPs were similarly monitored in the presence or absence of DTT.
In addition, to evaluate the reduction-sensitive MW change of mPEG-RSPCL copolymers as a component of mPEG-RSPCL NPs, the NPs (0.25 mg mL−1) were exposed to 10 mM DTT in DPBS (1 mL, 50 mM, pH 7.4). To measure the MW of mPEG-RSPCL polymer fragments, the aqueous solution of the polymer and its fragments was lyophilized to remove water and the dried powder was dissolved in THF. After filtering out the undissolved chemicals (e.g., salts), the solution was analyzed by GPC using a PLgel column (5 μm Mixed-D 300 × 7.5 column; Agilent Technologies, Santa Clara, CA, USA) and THF as the eluent at a flow rate of 1.0 mL min−1 at 35 °C. The MW, MWD, and PDI of the degraded mPEG-RSPCL fragments were analyzed by comparison with a MW standard curve of monodisperse polystyrenes.
Preparation and physicochemical characterization of drug-loaded mPEG-RSPCL NPs
This study used water-soluble DOX·HCl and the salt-free, water-insoluble DOX as model drugs. In feed, the target concentration of NPs was 0.5 mg mL−1 and the target loading content of model drug in NPs was 10 wt%. First, to load the relatively hydrophilic DOX·HCl into mPEG-RSPCL NPs, a drug loading method using an AS gradient was applied after preparing the drug-free mPEG-RSPCL NPs. In brief, after adding mPEG-RSPCL copolymers (2 mg) dissolved in DMSO (100 μL) into DIW (3.856 mL), the formed mPEG-RSPCL NP solution was stirred for 30 min and then dialyzed against DIW using a dialysis membrane (MWCO 2 kDa) to remove DMSO. AS powder was dissolved in the NP-containing solution at a concentration of AS 250 mM. Then, DOX·HCl in DIW (0.22 mg in 44.4 μL) was added into the NP solution with AS and stirred for 2 h at 60 °C. For evaluating effects of temperature in loading efficiency of DOX·HCl in mPEG-RSPCL NPs, 20 °C or 40 °C were also applied. Finally, DOX·HCl-loaded mPEG-RSPCL (DOX·HCl-mPEG-RSPCL) NPs were dialyzed against DIW using a dialysis membrane (MWCO 15 kDa) at 4 °C to remove unloaded DOX·HCl.Second, relatively hydrophobic DOX was loaded into mPEG-RSPCL NPs. After DOX·HCl and TEA (3 equivalents of DOX·HCl) were incubated in DMSO overnight, HCl was detached from DOX·HCl and the resulting water-insoluble DOX was obtained. A mixed solution of DOX (0.22 mg) and mPEG-RSPCL copolymers (2 mg) in DMSO (0.144 mL) was slowly injected into DIW (3.856 mL) and then stirred vigorously for 1 h. The DOX-mPEG-RSPCL NP solution was transferred into a dialysis membrane (MWCO 15 kDa) and dialyzed against DIW at 4 °C to remove DMSO. The drug-loaded NP solution was filtered by a 0.45 μm syringe filter to remove unloaded DOX.
To evaluate the drug loading content (DLC) and drug loading efficiency (DLE) in mPEG-RSPCL NPs, the prepared DOX·HCl-mPEG-RSPCL NPs and DOX-mPEG-RSPCL NPs were lyophilized and then dissolved in DMSO. After monitoring absorbance of drugs loaded in the NPs at 485 nm with a microplate reader (SpectraMax M5, Molecular devices, Sunnyvale, CA), their concentrations were estimated by a concentration (CONC)-absorbance (ABS) standard curve of either DOX·HCl prepared in DMSO (CONCDOX·HCl (μg mL−1) = 208.81ABSDOX·HCl − 0.2871; r2 ≈ 0.9996) or DOX prepared in DMSO (CONCDOX (μg mL−1) = 0.1957ABSDOX − 0.0003; r2 ≈ 0.9999) DLC and DLE were calculated using the following equations:


For comparison, the DLC and DLE of the control NPs were similarly evaluated.
Morphological evaluation of drug-loaded mPEG-RSPCL NPs
To evaluate the morphology of drug-loaded mPEG-RSPCL NPs, cryogenic transmission electron microscopy (cryo TEM) experiments were performed with a thin film of the NP solution (4 μL, 0.5 mg mL−1) transferred onto a lacey supported grid. The thin aqueous films were prepared at ambient temperature in a humidity of 97–99% within a custom-built environmental chamber to prevent evaporation of water from the sample solution. The excess liquid was blotted with a filter paper for 2–3 s, and the thin aqueous films were rapidly vitrified by plunging them into liquid ethane (cooled by liquid nitrogen) at its freezing point. The prepared samples of NPs were transferred to a Gatan 626 cryo holder. The samples were observed using a JEM-3010 HR at liquid nitrogen temperature to prevent sublimation with a 120 kV accelerating voltage, and the images were acquired with an SC 1000 CCD camera (Gatan, Inc.; Warrendale, PA). For comparison, the morphology of drug-free mPEG-RSPCL NPs was also monitored.Thiol-triggering drug release from drug-loaded mPEG-RSPCL NPs
DOX·HCl-mPEG-PCL NPs ([DOX·HCl] = 10 μg mL−1, 1 mL) in a floating dialysis cup (MWCO 3.5 kDa) were exposed to DTT-free or DTT (10 mM)-containing DPBS (50 mM, 4 mL, pH 7.4) at 37 °C. Then, the release experiments were performed at 80 rpm and 37 °C. At predetermined time points, the solution in the receiver was replaced with DTT-free DPBS, and the dialysate was lyophilized. After the lyophilized powder was dissolved in DMSO, the DOX·HCl concentration was estimated by observing its fluorescence at 479 nm (excitation) and 593 nm (emission). The % drug release was calculated by the following equation.
For comparison, the DOX·HCl release from the control NPs was similarly evaluated.
Cytotoxicity and in vitro anti-tumor efficacy of drug-free mPEG-RSPCL NPs and drug-loaded mPEG-RSPCL NPs
Using a well-known MTT assay, material cytotoxicity of drug-free mPEG-RSPCL NPs ([NP] ≤ 400 μg mL−1) and in vitro anti-tumor effects of drug-loaded mPEG-RSPCL NPs ([drug] ≤ 10 μg mL−1) were evaluated in HeLa (human cervical adenocarcinoma cell lines) and HepG2 (human hepatoma cell lines) cells. HeLa and HepG2 cells were cultured in RPMI-1640 and DMEM, respectively, supplemented with 10% FBS and D-glucose (2 g L−1 for RPMI-1640 and 4.5 g L−1 for DMEM) under humidified air with 5% CO2 at 37 °C. Cells were seeded in 96-well plates at a density of 2000 cells per well for HeLa cells and a density of 5000 cells per well for HepG2 cells in culture medium (0.1 mL) and were incubated for 24 h. After treated with free drugs, drug-free NPs, or drug-loaded NPs, the cells were incubated for 48 h and then MTT solution (10 μL, 5 mg mL−1) was added into the cells. After additional 4 h incubation, the MTT-containing culture medium was discarded and DMSO (0.1 mL) was added to dissolve formazan crystals produced by live cells. The absorbance of the formazan was measured at 570 nm using a microplate reader (SpectraMax M5; Molecular Devices, Sunnyvale, CA) and used to calculate viabilities of free drug-treated or NP-treated cells by the following equation. Drug-free mPEG-RSPCL NPs were only treated to evaluate their material cytotoxicity, whereas in vitro anti-tumor efficacies of DOX·HCl-mPEG-RSPCL NPs and DOX-mPEG-RSPCL NPs were compared with those of free DOX·HCl and free DOX.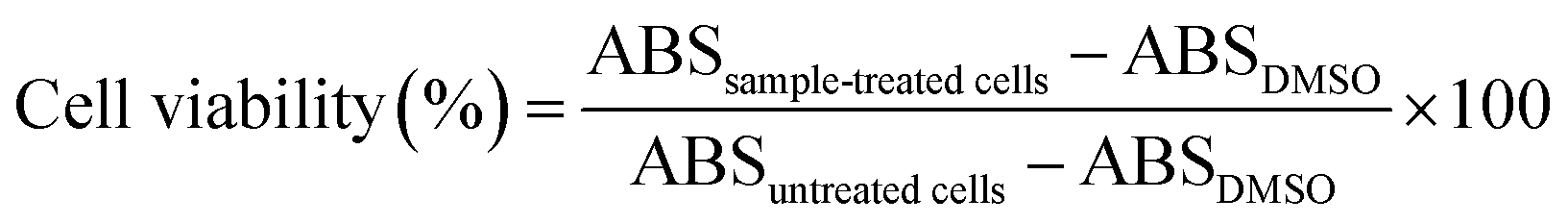
For comparison, the cell viability of the drug-free and drug-loaded control NPs was similarly evaluated.
Cellular uptake and intracellular localization of drug-loaded mPEG-RSPCL NPs
To assess cellular uptakes of free drugs or drug-loaded mPEG-RSPCL NPs, cells were seeded in 6-well plates at a density of 5 × 105 cells per well in the culture medium (2 mL) and were incubated for 24 h. After a 4 h treatment with free drugs, DOX·HCl-mPEG-RSPCL NPs, or DOX-mPEG-RSPCL NPs ([DOX] = 5 μg mL−1), the cells were rinsed twice with DPBS and detached. Cellular DOX fluorescence was measured using a flow cytometer (FACScanto II, Becton–Dickinson, Franklin Lakes, NJ, USA) with a primary argon laser (532 nm) and a fluorescence detector (578 ± 15 nm). The DOX uptake in the cells was analyzed from the gated viable population of at least 5 × 103 cells. To compare cellular uptakes of samples (i.e., free drugs and drug-loaded NPs), their normalized cellular uptakes were calculated as the following equation:
To measure intracellular localization of the drugs delivered with drug-loaded mPEG-RSPCL NPs ([DOX] = 5 μg mL−1), cells (3 × 104 cells per well) were seeded in a confocal dish and incubated for 24 h. Before treatment with drug-loaded mPEG-RSPCL NPs, the culture medium was replaced with fresh medium (0.3 mL). After a 4 h treatment of drug-loaded mPEG-RSPCL NPs, the culture medium was replaced. Also, after adding Hoechst33342 (10 μg mL−1) for nuclei staining in the culture medium, the cells were incubated for additional 10 min. The cells were rinsed with DPBS for further evaluation with a laser scanning confocal microscope (LSM710; Carl Zeiss, Oberkochen, Germany) equipped with excitation lasers (408 nm for diode and 543 nm for HeNe) and variable band-pass emission filters.
Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF) grants funded by the Korea government (MSIP) (NRF-2015R1A1A05001459 and NRF-2013R1A2A2A04015914) and Research Fund of The Catholic University of Korea (2015).Notes and references
- B. Lindman and P. Alexandridis, in Amphiphilic block copolymers: Self-assembly and applications, ed. P. Alexandridis and B. Lindman, Elsevier, Amsterdam, 2000, pp. 1–12 Search PubMed
.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed
- T. Smart, H. Lomas, M. Massignani, M. V. Flores-Merino, L. R. Perez and G. Battaglia, Nano Today, 2008, 3, 38–46 CrossRef CAS
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343–1355 CrossRef CAS PubMed
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed
- R. Duncan and M. J. Vicent, Adv. Drug Delivery Rev., 2013, 65, 60–70 CrossRef CAS PubMed
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed
- L. L. Ma, P. Jie and S. S. Venkatraman, Adv. Funct. Mater., 2008, 18, 716–725 CrossRef CAS
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed. Engl., 2010, 49, 6288–6308 CrossRef CAS PubMed
- T. K. Dash and V. B. Konkimalla, Mol. Pharm., 2012, 9, 2365–2379 CrossRef CAS PubMed
- H. Seyednejad, A. H. Ghassemi, C. F. van Nostrum, T. Vermonden and W. E. Hennink, J. Controlled Release, 2011, 152, 168–176 CrossRef CAS PubMed
- K. Yoon, H. C. Kang, L. Li, H. Cho, M.-K. Park, E. Lee, Y. H. Bae and K. M. Huh, Polym. Chem., 2015, 6, 531–542 RSC
- L. Li, H. Cho, K. H. Yoon, H. C. Kang and K. M. Huh, Int. J. Pharm., 2014, 471, 339–348 CrossRef CAS PubMed
- H. Sah, L. A. Thoma, H. R. Desu, E. Sah and G. C. Wood, Int. J. Nanomed., 2013, 8, 747–765 CrossRef PubMed
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed
- Y. Xiao, M. Yuan, J. Zhang, J. Yan and M. Lang, Curr. Top. Med. Chem., 2014, 14, 781–818 CrossRef CAS PubMed
- H. C. Kang, E. S. Lee, K. Na and Y. H. Bae, in Multifunctional Pharmaceutical Nanocarriers, ed. V. P. Torchilin, Springer, New York, 2008, vol. 4, pp. 161–199 Search PubMed
- H. Sun, F. Meng, R. Cheng, C. Deng and Z. Zhong, Expert Opin. Drug Delivery, 2013, 10, 1109–1122 CrossRef CAS PubMed
- H. C. Kang, O. Samsonova, S. W. Kang and Y. H. Bae, Biomaterials, 2012, 33, 1651–1662 CrossRef CAS PubMed
- H. C. Kang, K. M. Huh and Y. H. Bae, J. Controlled Release, 2012, 164, 256–264 CrossRef CAS PubMed
- H. C. Kang and Y. H. Bae, in Organelle-specific pharmaceutical nanotechnology, ed. V. Weissig and G. G. M. D'Souza, John Wiley & Sons, Inc., Hoboken, NJ, 2010, ch. 14, pp. 247–262 Search PubMed
- H. Cho, Y. Y. Cho, Y. H. Bae and H. C. Kang, Adv. Healthcare Mater., 2014, 3, 1007–1014 CrossRef CAS PubMed
- H. C. Kang and Y. H. Bae, Adv. Funct. Mater., 2007, 17, 1263–1272 CrossRef CAS
- L. Tian, H. C. Kang and Y. H. Bae, Biomacromolecules, 2013, 14, 2570–2581 CrossRef CAS PubMed
- H. C. Kang, H. J. Kang and Y. H. Bae, Biomaterials, 2011, 32, 1193–1203 CrossRef CAS PubMed
- H. S. Hwang, H. C. Kang and Y. H. Bae, Biomacromolecules, 2013, 14, 548–556 CrossRef CAS PubMed
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS PubMed
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS PubMed
- W. Chen, M. Zheng, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Biomacromolecules, 2013, 14, 1214–1222 CrossRef CAS PubMed
- J. Liu, Y. Pang, W. Huang, X. Huang, L. Meng, X. Zhu, Y. Zhou and D. Yan, Biomacromolecules, 2011, 12, 1567–1577 CrossRef CAS PubMed
- Y. Zhong, W. Yang, H. Sun, R. Cheng, F. Meng, C. Deng and Z. Zhong, Biomacromolecules, 2013, 14, 3723–3730 CrossRef CAS PubMed
- S. Samarajeewa, R. Shrestha, M. Elsabahy, A. Karwa, A. Li, R. P. Zentay, J. G. Kostelc, R. B. Dorshow and K. L. Wooley, Mol. Pharm., 2013, 10, 1092–1099 CrossRef CAS PubMed
- X. Zhao and P. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 166–174 CAS
- S. Cajot, D. Schol, F. Danhier, V. Preat, M. C. Gillet De Pauw and C. Jerome, Macromol. Biosci., 2013, 13, 1661–1670 CrossRef CAS PubMed
- A. Kumar, S. V. Lale, S. Mahajan, V. Choudhary and V. Koul, ACS Appl. Mater. Interfaces, 2015, 7, 9211–9227 CAS
- D. Basak, R. Bej and S. Ghosh, Polym. Chem., 2015, 6, 6465–6474 RSC
- D. E. Discher, V. Ortiz, G. Srinivas, M. L. Klein, Y. Kim, D. Christian, S. Cai, P. Photos and F. Ahmed, Prog. Polym. Sci., 2007, 32, 838–857 CrossRef CAS PubMed
- D. Y. Cho, H. Cho, K. Kwon, M. Yu, E. Lee, K. M. Huh, D. H. Lee and H. C. Kang, Adv. Funct. Mater., 2015, 25, 5479–5491 CrossRef CAS
- K. Kataoka, T. Matsumoto, M. Yokoyama, T. Okano, Y. Sakurai, S. Fukushima, K. Okamoto and G. S. Kwon, J. Controlled Release, 2000, 64, 143–153 CrossRef CAS PubMed
- P. Changenet-Barret, T. Gustavsson, D. Markovitsi, I. Manet and S. Monti, Phys. Chem. Chem. Phys., 2013, 15, 2937–2944 RSC
- W. L. Kim, H. Cho, L. Li, H. C. Kang and K. M. Huh, Biomacromolecules, 2014, 15, 2224–2234 CrossRef CAS PubMed
- H. Yin, H. C. Kang, K. M. Huh and Y. H. Bae, Colloids Surf., B, 2014, 116, 128–137 CrossRef CAS PubMed
- J. Xu, Q. Zhao, Y. Jin and L. Qiu, Nanomedicine, 2014, 10, 349–358 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra25319e |
| ‡ SYM and YSC equally contributed this work. |
| This journal is © The Royal Society of Chemistry 2016 |