
Amorphous apatite thin film formation on a biodegradable Mg alloy for bone regeneration: strategy, characterization, biodegradation, and in vitro cell study†
Abstract
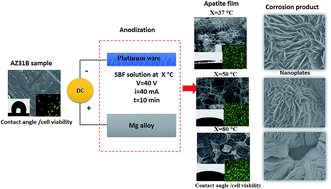
Bioactive films with a nanoplate structure were prepared on the surface of a biodegradable AZ31B magnesium (Mg) alloy via anodization in simulated body fluid (SBF) as an electrolyte to control Mg biodegradability and improve surface bioactivity. The effect of the electrolyte temperature and pH values on the formation of the biomimetic film were studied. The electrolyte was set at three different temperatures of 37, 50, and 80 °C, with pH values ranging from 7.4 to 8 for the lower electrolyte temperature and 11.5–12 for the two higher levels of temperature. The apatite films on the different samples were characterized using X-ray diffraction spectroscopy (XRD), field emission scanning electron microscopy (FE-SEM), EDS element mapping, X-ray photon spectroscopy (XPS), and FTIR spectroscopy. The water contact angles of the different surfaces were evaluated, moreover, the corrosion behaviors of the different samples were studied using electrochemical potentiodynamic DC, electrochemical impedance spectroscopy (EIS), and immersion tests. The human fetal-osteoblast cell line hFOB 1.19 was used in a cell culture test, and the biological response and cell function were evaluated in vitro using DNA and PCR. The decomposition of the apatite film was affected by the anodization electrolyte temperature, resulting in an amorphous structure. It is observed that the apatite structure has nanoplates at electrolyte temperature (37 to 50) °C and these have a tendency to disappear at 80 °C.
 Please wait while we load your content...
Please wait while we load your content...

