
Mn-doped La0.6Sr0.4CoO3 perovskite catalysts with enhanced performances for non-aqueous electrolyte Li–O2 batteries
Nian Sun,
Hanxing Liu*,
Zhiyong Yu,
Zhenning Zheng and
Chongyang Shao
State Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Hubei Province 430070, PR China. E-mail: lhxhp@whut.edu.cn; Fax: +86 27 87885813; Tel: +86 27 87653330
First published on 25th January 2016
Abstract
Mn-doped perovskite oxides, La0.6Sr0.4Co1−xMnxO3 (x = 0.05, 0.1), have been synthesized and Li–O2 batteries based on La0.6Sr0.4Co1−xMnxO3/Super P electrodes and non-aqueous electrolytes were also fabricated and measured in this work. The oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) activity of the as-prepared electrode based Li–O2 batteries were measured using cyclic voltammetry (CV) tests. In terms of the ORR and OER peak current, the performance is enhanced in the order of pure Super P, La0.6Sr0.4CoO3, La0.6Sr0.4Co0.95Mn0.05O3 and La0.6Sr0.4Co0.9Mn0.1O3. The Li–O2 batteries based on a La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode exhibited a higher first discharge capacity, lower over-potential and better cycling stability, which was in agreement with the trend shown in the CV curves. The electrochemical impedance spectroscopy measurements revealed that the impedance of the battery increased slowly during cycling due to the accumulation of undecomposed discharge products on the cathode.
1. Introduction
Li–air batteries, generally also called Li–O2 batteries have become a focus of current research interests due to their extremely high theoretical energy density of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 W h kg−1, which is a much higher capacity than that of today’s Li-ion batteries.1–6 Therefore they are considered as one of the potential substitutes of gasoline for application in electric vehicles to satisfy the energy requirement in the near future.1,2,6 In a typical non-aqueous electrolyte Li–O2 battery system, the cell is composed of a lithium metal anode, a separator, a Li+ conducting electrolyte and a porous air electrode based on the carbon matrix and catalyst, while the basic chemical reactions during the discharge–charge process involve the formation of Li oxides (Li2O2 or Li2O) and the decomposition of them. However, the practical power density is much lower than the theoretical predication and the rate of these reactions is too sluggish to satisfy the requirements for practical application because of the low oxygen solubility in non-aqueous electrolytes and oxygen mobility.7,8 At present, Li–O2 batteries still suffer from some significant limitations such as poor cycle life, low rate capability, instability of the electrolyte and a high over-potential.9–11 In fact, to overcome the above mentioned disadvantages strongly depending on the air electrode, catalysts with high activity are utilized in the cathode to promote the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) kinetics, which has become the key focus of many researchers.9,12–14
000 W h kg−1, which is a much higher capacity than that of today’s Li-ion batteries.1–6 Therefore they are considered as one of the potential substitutes of gasoline for application in electric vehicles to satisfy the energy requirement in the near future.1,2,6 In a typical non-aqueous electrolyte Li–O2 battery system, the cell is composed of a lithium metal anode, a separator, a Li+ conducting electrolyte and a porous air electrode based on the carbon matrix and catalyst, while the basic chemical reactions during the discharge–charge process involve the formation of Li oxides (Li2O2 or Li2O) and the decomposition of them. However, the practical power density is much lower than the theoretical predication and the rate of these reactions is too sluggish to satisfy the requirements for practical application because of the low oxygen solubility in non-aqueous electrolytes and oxygen mobility.7,8 At present, Li–O2 batteries still suffer from some significant limitations such as poor cycle life, low rate capability, instability of the electrolyte and a high over-potential.9–11 In fact, to overcome the above mentioned disadvantages strongly depending on the air electrode, catalysts with high activity are utilized in the cathode to promote the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) kinetics, which has become the key focus of many researchers.9,12–14
The products of the cell reactions including Li2O and Li2O2 are not soluble in organic electrolyte solutions and these oxides grow in the pores of the carbon matrix and even the current collector,15 which blocks further transportation of oxygen, makes charge transfer more difficult and finally obstructs the discharge of battery. The highly efficient cathode catalysts are able to improve the discharge and charge process of the cell by optimizing the accumulation of discharge products on the air electrode surface and facilitating the decomposition of discharge products,9,16 so the adoption of an ideal catalyst in Li–O2 batteries could affect the specific capacity, over-potential, energy efficiency, and cycle performance somewhat.17 Perovskite oxides, having a formula of ABO3, are widely interesting cathode catalysts in solid oxide fuel cells due to their oxygen vacancy and excellent oxygen mobility.18 Many researchers18–21 have demonstrated that by means of partially substituting a proper cation A′ for A, or a cation B′ for B, with a different valence, an increase in oxygen vacancy concentration and changes in the valence states of the active metal B would take place in the perovskite material therefore influencing its catalytic activity and conductivity. La0.6Sr0.4CoO322,23 with the perovskite structure have been considered as a potential catalyst for high performance Li–O2 batteries because of high oxygen ionic conductivity, good oxygen permeability and high catalytic activity for oxidation. Li8 and his colleagues have demonstrated that one-dimensional porous La0.5Sr0.5CoO2.91 nanotubes could promote both the oxygen reduction and oxygen evolution reactions. In addition, Li–O2 batteries on the basis of that catalyst and non-aqueous electrolytes displayed a very high initial discharge capacity of 7205 mA h g−1. For the perovskite oxides, the thermal stability of materials with Mn3+ in the B site is better than that of materials with Co3+ in the B site. Moreover, La1−xSrxMnO3 perovskites have been reported as an effectively bifunctional catalyst in Li–O2 batteries and demonstrated promising catalytic properties. For example, Lu et al.24 proposed microporous La0.8Sr0.2MnO3 perovskite nanorods as a highly active electrocatalyst fabricated via a soft template method for Li–O2 batteries, which displayed a high first discharge specific capacity (6890 mA h g−1), low over-potential and good rate capability. In our previous work,25 we fabricated and characterized Li–O2 batteries on the basis of a La0.6Sr0.4CoO3/Super P electrode, which displayed good catalytic performance. Herein, we suppose to promote the catalytic performance of La0.6Sr0.4CoO3 perovskite materials by means of doping Mn in the B site to partially substitute for Co3+ because Mn3+ in the B site could make this perovskite oxide more stable and its catalytic activity is also excellent.
In this work, we have synthesized Mn doped La0.6Sr0.4CoO3 nanoparticles by using a dry gel combustion method. The prepared samples displayed a loose structure. The Li–O2 batteries based on the catalyst have been also fabricated and characterized. The results indicated that Mn doped La0.6Sr0.4CoO3 nanoparticles as a catalyst showed an enhanced specific discharge capacity, cycle stability and lower over-potential compared with a La0.6Sr0.4CoO3 nanoparticle catalyst.
2. Experimental section
2.1 Synthesis of La0.6Sr0.4Co1−xMnxO3 powder
The raw materials used in this experiment were of analytical grade and purchased from Guoyao Chemical Reagent Co. Ltd.All the perovskite oxide nanopowders were synthesized by using the dry-gel combustion method. The stoichiometric ratio of La(NO3)3·6H2O, Sr(NO3)2, Co(NO3)2·6H2O and Mn(NO3)2 were dissolved in deionized water, respectively. Subsequently, a certain amount of citric acid and EDTA (ethylene diamine tetraacetic acid, dissolved in moderate NH4OH solution at first) were added into the mixed solution followed by adding necessary NH4OH to maintain the pH between 8 and 9. The mole ratio of total metal ions to EDTA and to citric acid was 1:1:1.5.22 Then the precursor solution was kept at a high stirring rate at 80 °C for 6 h and the final the dry gel formed at 120 °C for one night in a draught drying cabinet. The dry gel was completely burned in muffle at 500 °C for about 15 min and then ground before being calcined in air at 900 °C (heating rate of 3 °C min−1) for 6 h to obtain the final perovskite powders.
2.2 Material characterization
The phase structures of the La0.6Sr0.4Co1−xMnxO3 product were analyzed using X-ray diffraction (X’Pert, PANalytical, Netherlands) with the parameters of Cu target (λ = 1.54056 Å), 40 kV working voltage, 40 mA working current and a 2θ scanning range from 20° to 80°. The micromorphology of the product was observed using field emission scanning electron microscopy (FESEM, S-4800, Japan). The accelerating voltage was 20 kV and the enlargement factor was between 30 and 800000 times. The valence state of the elements in the perovskite oxides were obtained by using X-ray photoelectron spectroscopy (XPS. VG Multilab 2000).
2.3 Electrode fabrication and battery measurement
The carbon (Super P) and La0.6Sr0.4Co1−xMnxO3 were homogeneously mixed before preparing the catalyst ink. The moderate Super P and La0.6Sr0.4Co1−xMnxO3 were ultrasonically dispersed in alcohol for 2 h, followed by consistent high speed rate stirring at room temperature for 4 h. The final black mixtures were obtained after evaporation of the alcohol at 100 °C. The cathode ink contained a carbon matrix (Super P), the catalyst (La0.6Sr0.4Co1−xMnxO3) and a binder (polyvinylidene fluoride, PVDF). The mass ratio of catalyst:carbon:binder was adjusted to 27:63:10. These components were stirred in a moderate 1-methyl-2-pyrrolidone solution for at least 24 h for homogeneous mixing, then they were coated on Ni mesh with a diameter of 14 mm and dried at 80 °C in air for 1 h and 120 °C in a vacuum oven for 12 h. The electrode loading was controlled at around 0.65 mg cm−2.
The composition of one battery is shown in Fig. 1. The Li–O2 battery is composed of La0.6Sr0.4Co1−xMnxO3/Super P as the cathode, a lithium wafer as the anode, one layer of glass fiber as the separator (Whatman, GF/D), and 1 M solution of LiTFSI (bis(trifluoromethane)sulfonimide lithium salt) in tetramethylene sulfone as the electrolyte. The batteries were assembled by using a stainless steel mould in an Ar-filled glove box with <0.01 ppm O2 and moisture content. For fabricating a battery, the bottom current collector, lithium wafers, one layer of separator, Ni mesh coated with La0.6Sr0.4Co1−xMnxO3/Super P and the top current collector were stacked layer by layer as shown in the left schematic diagram. Then they were placed on the bottom of the battery mould and covered by the PTFE insulation sleeve with a compression spring. Finally the top of the battery mould was used to cover them as shown in the middle schematic diagram. The O2 was injected into the battery mould from the entrance of one side and held on through closing the two gas checks. Then measurements could be carried out by connecting the wires with metal sticks on the mould.
 | ||
| Fig. 1 Schematic showing the internal structure of the Li–O2 battery and battery model and a photograph of the mould. | ||
The electrochemical performance of the batteries was recorded using a battery test system (LANHE-CT2001A). Electrochemical tests were carried out in the mould filled with O2 (pressure of 0.05 MPa). To determine the maximum capacity of the cell, parts of the batteries were set to a full discharge–charge with the voltage range of 2.0 to 4.5 V. The other batteries were limited at a capacity of 500 mA h g−1 at a current density of 100 mA g−1 to evaluate the cycle performance. The alternating-current impedance (AC impedance) in the frequency range 0.01–105 Hz and cyclic voltammetry (CV) curves were tested using an electrochemical workstation (CHI660B) to study the internal resistance change during the discharge–charge process and catalytic performance respectively.
3. Results and discussion
3.1 Materials characterization
Fig. 2 shows the XRD data of the La0.6Sr0.4Co1−xMnxO3 (x = 0, 0.05, 0.1, 0.15) powders calcined at 900 °C for 6 h in air. The data indicated that the as-prepared La0.6Sr0.4Co1−xMnxO3 perovskite oxides display consistent diffraction of La0.6Sr0.4CoO3 (JCPDS card no. 01-809-5719: a = 5.4159 Å, b = 5.4159 Å, c = 13.1920 Å). A small amount of Mn oxide existed in the doped samples with a high Mn doping amount of 15% while other impurity phases were not observed in any samples. The enlarged (104) peak was also given at the right of the figure and it gradually moved to the left with an increase in Mn content, indicating that the lattice constant increased. In order to eliminate the possible influence of Mn oxides, the La0.6Sr0.4Co0.95Mn0.05O3 and La0.6Sr0.4Co0.9Mn0.1O3 samples were selected to contrast with La0.6Sr0.4CoO3. | ||
| Fig. 2 XRD patterns of La0.6Sr0.4Co1−xMnxO3 (x = 0, 0.05, 0.1, 0.15) powders calcined at 900 °C in air for 6 h. | ||
Fig. 3(a) shows the microstructure of La0.6Sr0.4CoO3 magnified up to 3400 times. It shows clearly that the sample displayed a loose structure, which is attributed to the intense combustion of EDTA and citric acid at 500 °C in a few minutes. Fig. 3(b)–(d) show the microtopography of La0.6Sr0.4Co1−xMnxO3 (x = 0, 0.05, 0.1) samples magnified up to 30000 times. The BET surface areas of the samples was about 4.59 m2 g−1 and the grain size (100–120 nm) was hardly changed at all with the increase of Mn.
 | ||
| Fig. 3 SEM of (a) La0.6Sr0.4CoO3 sample magnified up to 3400 times, (b) La0.6Sr0.4CoO3 (c) La0.6Sr0.4Co0.95Mn0.05O3 and (d) La0.6Sr0.4Co0.9Mn0.1O3 samples magnified up to 30000 times. | ||
The chemical states and surface composition of Co, Mn and O in the La0.6Sr0.4Co1−xMnxO3 (x = 0, 0.05, 0.1) samples were measured using X-ray photoelectron spectroscopy (XPS). Fig. 4(a) shows the Co 2p XPS spectra of La0.6Sr0.4Co1−xMnxO3 perovskites. Two peaks at a higher binding energy (781.6 eV and 789.6 eV) could be assigned to Co2+ and one peak at a lower binding energy (779.9 eV) could be assigned for Co3+. The signal fraction of the species with lower oxidation state (Co2+) slightly increased with the replacement of Co by Mn.
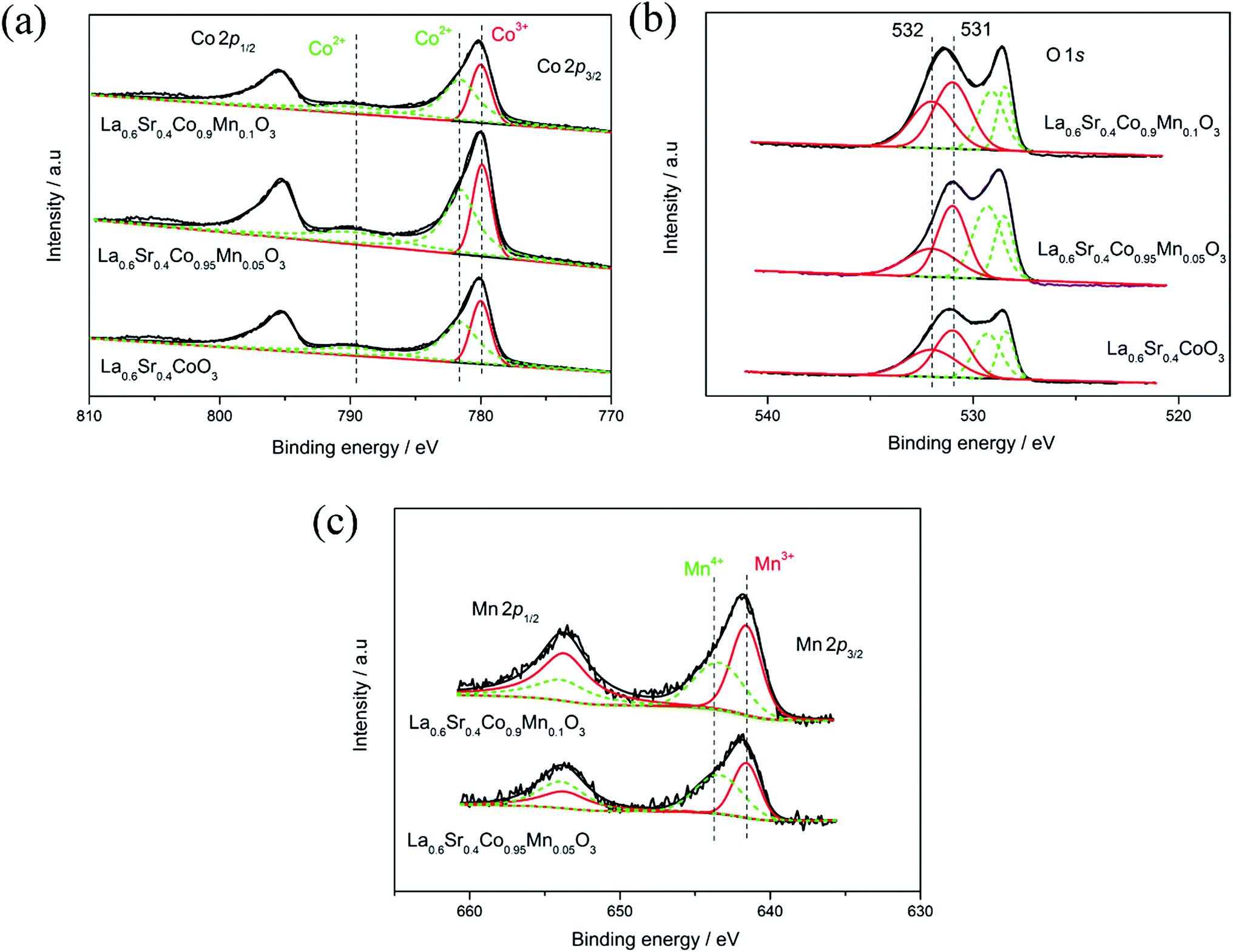 | ||
| Fig. 4 (a) Co 2p, (b) O 1s, and (c) Mn 2p spectra of La0.6Sr0.4Co1−xMnxO3 (x = 0, 0.05, 0.1) perovskite oxides. | ||
The XPS spectra of O 1s for the La0.6Sr0.4Co1−xMnxO3 (x = 0, 0.05, 0.1) samples are shown in Fig. 4(b). It can be concluded that the oxygen may exist as four different oxygen species. The first two peaks at the lower binding energy range (528.4–529.3 eV) were assigned to the lattice oxygen (O2−) and the other two peaks located at the higher binding energy range (531–532 eV) could be assigned to oxygen atoms in hydroxyl groups, which may be due to the oxygen atoms of surface hydroxyl groups.8,22,23 It could be found that the latter two peaks corresponding to the adsorbed oxygen species became stronger with the increase in Mn content, suggesting an improvement in the catalytic activities for the ORR and OER.26,27
Fig. 4(c) displays the Mn 2p XPS spectra of La0.6Sr0.4Co1−xMnxO3 (x = 0.05, 0.1) perovskites. Two Mn 2p2/3 peaks corresponding to different chemical states (643.4 eV typical for Mn4+, 641.6 eV typical for Mn3+) could be observed.
3.2 Electrochemical characterization
To study the catalytic performance of the as-prepared electrodes, CV curves for Li–O2 batteries were tested within the voltage range of 2.0 to 4.5 V, shown in Fig. 5(c). The CV curve of the La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode based Li–O2 battery exhibited higher peak currents and ORR onset potential (∼2.8 V) compared with the other electrode based Li–O2 batteries, indicating that both the ORR and OER were enhanced by La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode, which was beneficial to improve the discharge capacity, recharging characteristic and round-trip efficiency of Li–O2 batteries.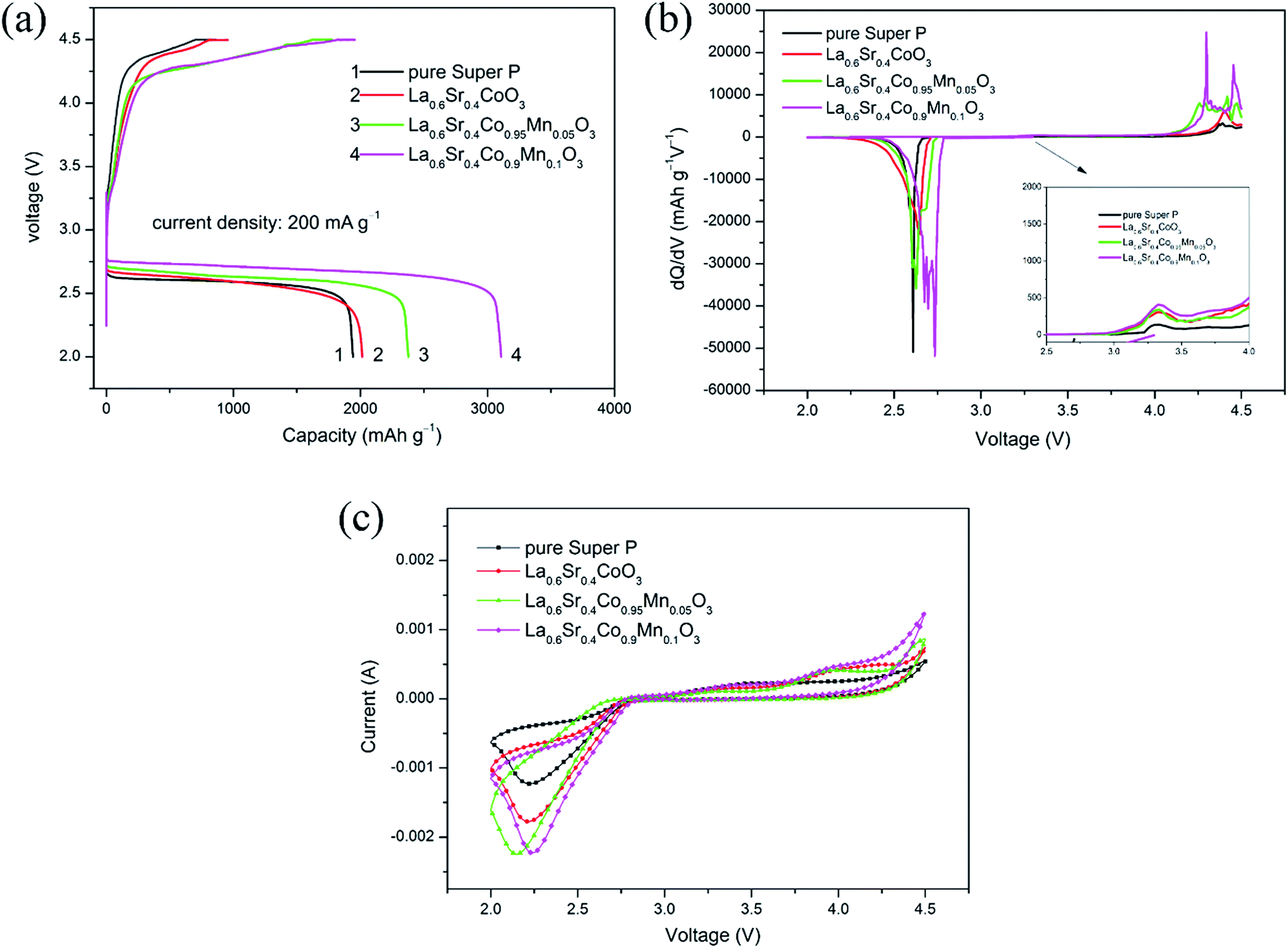 | ||
| Fig. 5 (a) Initial discharge–charge profiles of different Li–O2 batteries at 200 mA g−1; (b) the dQ/dV curves corresponding to different Li–O2 batteries at 200 mA g−1; (c) CV curves of different electrodes based Li–O2 batteries with a scan rate of 1 mV s−1. | ||
The electrochemical properties of all the batteries were examined by using the mould filled with O2. The first discharge specific capacity and cycle performance measurements were carried out in the voltage range of 2.0–4.5 V to avoid the possible decomposition of the electrolyte at the high charge voltage. For the purpose of providing enough energy for the decomposition of the reaction products, after the voltage reached 4.5 V during constant current charging, it was held at that value until the charge current was 1/20 of the discharge current during constant voltage charging. The capacity shown in this article was based on the total carbon/catalyst mass.
The initial discharge–charge profiles of the Li–O2 batteries based on different electrodes at the current density of 200 mA g−1 are displayed in Fig. 5(a). It is obvious that the Li–O2 battery based on La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode showed the highest first discharge capacity of 3107 mA h g−1, which was higher than the discharge capacities of the Li–O2 batteries based on La0.6Sr0.4Co0.95Mn0.05O3/Super P, La0.6Sr0.4CoO3/Super P and pure Super P electrodes, corresponding to 2378, 2015 and 1940 mA h g−1, respectively. It could be found that the discharge and charge voltage of Li–O2 batteries can be improved with the help of a La0.6Sr0.4Co0.9Mn0.1O3 catalyst. The discharge voltage of the Li–O2 battery based on La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode was higher than that based on the La0.6Sr0.4Co0.95Mn0.05O3/Super P electrode by about 60 mV, La0.6Sr0.4CoO3/Super P electrode by about 80 mV and pure Super P electrode by about 90 mV, in addition its averaged charge voltage was obviously lower than that of the Li–O2 batteries based on La0.6Sr0.4CoO3/Super P and pure Super P electrodes. Based on the differences in discharge–charge curves of these four electrodes, it can be concluded that the La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode showed the best catalytic activity.
The dQ/dV profiles of the Li–O2 batteries based on different electrodes in the first discharge–charge process under the current density of 200 mA g−1 are shown in Fig. 5(b). It can clearly be seen that there is one peak (2.6–2.8 V) in the discharge process and at least two peaks (3.0–3.5 V and 4.0–4.5 V) in the charge process, which was consistent with the ORR and OER peaks of the CV curves. It can also be found that the peak potential in the discharge process of the La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode was the highest, while the peak potential in the charge process of the La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode was the lowest.
The capacity-limited cycling method was used in this work to further study the stability of Li–O2 batteries based on different electrodes due to the capacity limit was beneficial to promote cycling performance. All the discharge capacity limit of 500 mA h g−1 tests were performed under 100 mA g−1 within the voltage range of 2.0 to 4.5 V. At the controlled max charge voltage of 4.5 V, charge capacity loss existed in each cycle (Table 1). As shown in Fig. 6(a), the discharge and charge voltage of La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode based Li–O2 batteries in the first cycle were ∼2.74 V and ∼4.00 V accompanied with a round-trip efficiency of 68.5%. Its average loss of charge capacity per cycle was approximately 2.1 mA h g−1. In addition, the circulation of the La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode based Li–O2 battery was maintained 50 times with 3.28% voltage loss, indicating it had a good cyclability.
| Electrode | Initial (mA h g−1) | Terminal (mA h g−1) | Cycle (times) | Loss per cycle (mA h g−1) |
|---|---|---|---|---|
| Pure Super P | 500 | 349 | 13 | 11.6 |
| La0.6Sr0.4CoO3 | 500 | 370 | 22 | 5.9 |
| La0.6Sr0.4Co0.9Mn0.1O3 | 500 | 389 | 53 | 2.1 |
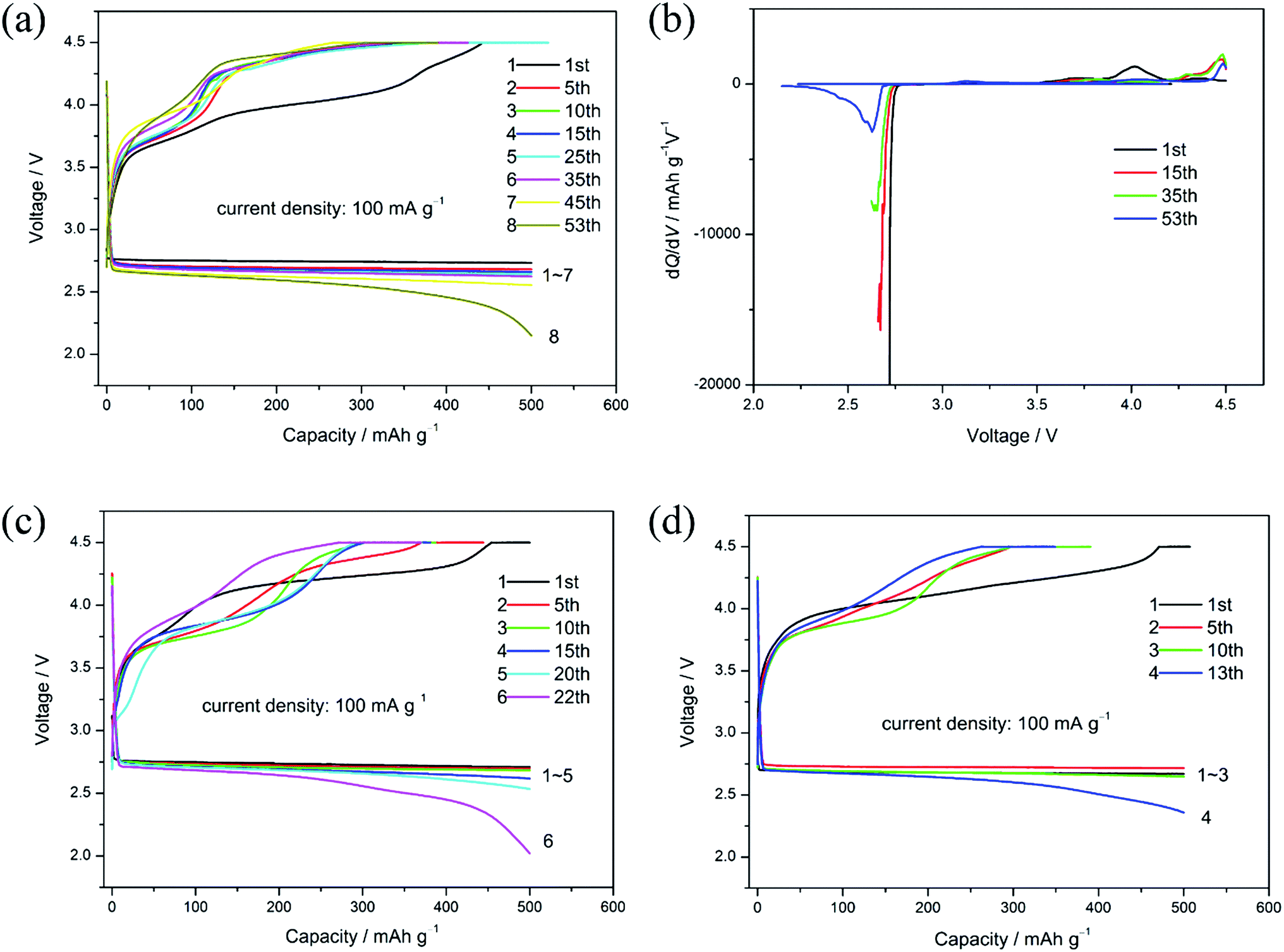 | ||
| Fig. 6 The cycling performance of La0.6Sr0.4Co0.9Mn0.1O3/Super P (a), La0.6Sr0.4CoO3/Super P (c), and the pure Super P (d) electrodes based Li–O2 batteries with a cut-off capacity of 500 mA h g−1 at 100 mA g−1; (b) different dQ/dV curves of the La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode based Li–O2 battery with a cut-off capacity of 500 mA h g−1 at 100 mA g−1. | ||
For comparison, the La0.6Sr0.4CoO3/Super P and pure Super P electrode based Li–O2 batteries were measured under similar experimental conditions. From Fig. 6(c) and (d), it can be seen that the circulation of La0.6Sr0.4CoO3/Super P and pure Super P electrodes based Li–O2 batteries decreased to only 22 and 13 cycles respectively under the same capacity constraint conditions. Moreover, their retention ability of charge capacity and round-trip efficiency became worse as well. The circulation ability decay may be ascribed to the catalytic activity of pure Super P and La0.6Sr0.4CoO3/Super P was lower than La0.6Sr0.4Co0.9Mn0.1O3/Super P indicated by the CV curves. Cathodes with better catalytic activity will promote the decomposition of discharge products (Li2O2), the accumulation of which on the electrode is the key factor leading to the serious deterioration of cell performance and further end of discharge. So, it can be concluded that the La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode had the best cycling stability according to the lower loss of charge capacity, longer cycle life and good discharge voltage maintaining ability.
The accumulation of discharge products such as Li2O2 and Li2O on the cathode surface will lead to an increase in resistance during the discharge process while these products may decompose during the charge process. To further investigate the discharge and charge behaviours, we studied the electrode composition after discharge and recharge using XRD. Under cell testing conditions of 100 mA g−1 current density and a 2.0–4.5 V voltage range, the crystalline Li2O2 (Fig. 7(a)) can be observed after the 1st discharge and then largely disappeared after the 1st recharge, indicating that parts of them had decomposed with the help of the catalyst. Obviously, the residual Li2O2 would accumulate on the electrode surface, leading to the decrease of electrode conductivity. Electrochemical impedance spectroscopy (EIS) data collected during the cycling process for Li–O2 batteries based on different electrodes are shown in Fig. 7(b)–(d). The AC-impedance spectra with one or two-arc behavior could be analyzed using the equivalent circuit in Fig. 7(e) or (f) respectively. According to the equivalent circuits Fig. 7(e) and (f), the ohmic resistance including the electrolyte, contact and cell resistance (represented by the high frequency intercept of the semi-circle on the real axis, Rs), the charge transfer resistance (represented by the middle frequency depressed semicircles, Rct) and Warburg resistance caused by diffusion (represented by the low frequency characteristic line, Ws) as a function of cycle processes were observed.28–30 The impedance of a constant phase element is given as follows:28

 | ||
| Fig. 7 (a) XRD patterns of pristine, the 1st-cycle discharged–recharged La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode with the voltage range of 2.0–4.5 V. The EIS comparison of the La0.6Sr0.4Co0.9Mn0.1O3/Super P (b), La0.6Sr0.4CoO3/Super P (c), and pure Super P (d) electrode based Li–O2 batteries with a limit capacity of 500 mA h g−1 at 100 mA g−1; (e) and (f) the equivalent circuits. | ||
| Different electrode based Li–O2 batteries | ||||
|---|---|---|---|---|
| Pure Super P | Cycles | 0 | 5 | 13 |
| Rct (Ω) | 32 | 62 | 94 | |
| La0.6Sr0.4CoO3 | Cycles | 0 | 5 | 10 | 15 | 21 |
| Rct (Ω) | 51 | 65 | 77 | 96 | 145 | |
| La0.6Sr0.4Co0.9Mn0.1O3 | Cycles | 0 | 5 | 10 | 15 | 25 | 35 | 53 |
| Rct (Ω) | 33 | 47 | 61 | 67 | 90 | 122 | 296 | |
4. Conclusions
Mn-doped La0.6Sr0.4CoO3 perovskite oxides were synthesized and identified as a catalyst for Li–O2 batteries using non-aqueous electrolyte. The CV curves of Li–O2 batteries on the basis of different electrodes revealed that Mn-doped perovskites La0.6Sr0.4Co1−xMnxO3 with higher peak current were higher-performance catalysts for both the ORR and OER. Besides, the initial discharge specific capacity of Li–O2 batteries based on La0.6Sr0.4Co1−xMnxO3/Super P and pure Super P electrodes were found to increase in the following order: Super P < La0.6Sr0.4CoO3 < La0.6Sr0.4Co0.95Mn0.05O3 < La0.6Sr0.4Co0.9Mn0.1O3, which was in good agreement to the trend of catalytic activity in the CV curves. The cycling performance test demonstrated good stability of the La0.6Sr0.4Co0.9Mn0.1O3/Super P electrode. The electrochemical impedance spectroscopy measurements indicated that the increase in impedance caused by the accumulation of discharge products on the cathode surface during cycling process was a significant factor leading to the end of discharge.Acknowledgements
This work was supported by the Natural Science Foundation of China (No. 51372191, No. 51102189), the International Science and Technology Cooperation Program of China (2011DFA52680), the program for New Century Excellent Talents in University (No. NCET-11-0685) the Key program of Natural Science Foundation of China (No. 50932004) and the Fundamental Research Funds for the Central Universities (WUT:2014-IV-134).References
- J. Christensen, P. Albertus, R. S. Sanchez-Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, J. Electrochem. Soc., 2012, 159, R1–R30 CrossRef CAS.
- G. B. Peter, A. F. Stefan, J. H. Laurence and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 Search PubMed.
- M. M. Thackeray, C. Wolverton and E. D. Isaac, Energy Environ. Sci., 2012, 5, 7854–7863 CAS.
- Y. Y. Shao, F. Ding, J. Xiao, J. Zhang, W. Xu, S. Park, J. G. Zhang, Y. Wang and J. Liu, Adv. Funct. Mater., 2012, 1–16 Search PubMed.
- P. P. R. M. L. Harks, F. M. Mulder and P. H. L. Notten, J. Power Sources, 2015, 288, 92–105 CrossRef CAS.
- A. Kraytsberg and E. Y. Ein, J. Power Sources, 2011, 196, 886–893 CrossRef CAS.
- Y. H. Chen, S. A. Freunberger, Z. Q. Peng, O. Fontaine and P. G. Bruce, Nat. Chem., 2013, 5, 489–494 CrossRef CAS PubMed.
- P. F. Li, J. K. Zhang, Q. L. Yu, J. S. Qiao, Z. H. Wang, D. Rooney, S. Sun and K. N. Sun, Electrochim. Acta, 2015, 165, 78–84 CrossRef CAS.
- A. Kraytsberg and E. Y. Ein, Nano Energy, 2013, 2, 468–480 CrossRef CAS.
- Y. Y. Shao, S. Park, J. Xiao, J. G. Zhang, Y. Wang and J. Liu, ASC Catal., 2012, 2, 844–857 CAS.
- D. Capsoni, M. Bini, S. Ferrari, E. Quartarone and P. Mustarelli, J. Power Sources, 2012, 220, 253–263 CrossRef.
- R. Mi, H. Liu, H. Wang, K. W. Wong, J. Mei, Y. G. Chen, W. M. Lau and H. Yan, Carbon, 2014, 67, 744–752 CrossRef CAS.
- H. Cheng and K. Scott, Appl. Catal., B, 2011, 108, 140–151 CrossRef.
- B. K. Ko, M. K. Kim, S. H. Kim, M. A. Lee, S. E. Shim and S. H. Baeck, J. Mol. Catal. A: Chem., 2013, 379, 9–14 CrossRef CAS.
- P. Tan, W. Shyy, T. S. Zhao, Z. H. Wei and L. An, J. Power Sources, 2015, 278, 133–140 CrossRef CAS.
- Q. Wang, D. Zheng, M. E. McKinnon, X. Q. Yang and D. Y. Qu, J. Power Sources, 2015, 274, 1005–1008 CrossRef.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- M. Neelima, B. Amitava, G. Alka, O. Shobit and B. Kantesh, Prog. Mater. Sci., 2015, 72, 141–337 CrossRef.
- Z. Z. Du, P. Yang, L. Wang, Y. H. Lu, J. B. Goodenough, J. Zhang and D. W. Zhang, J. Power Sources, 2014, 265, 91–96 CrossRef CAS.
- C. Jin, Z. B. Yang, X. C. Cao, F. L. Lu and R. Z. Yang, Int. J. Hydrogen Energy, 2014, 39, 2526–2530 CrossRef CAS.
- Y. F. Sun, N. Yan, J. H. Li, H. Y. Wu, J. L. Luo and K. T. Chuang, Sustainable Energy Technologies and Assessments, 2014, 8, 92–98 CrossRef.
- D. H. Prasad, S. Y. Park and E. O. Oh, Appl. Catal., A, 2012, 447–448, 100–106 CrossRef CAS.
- J. G. Deng, L. Zhang, H. X. Dai, H. He and C. T. Au, Catal. Lett., 2008, 123, 294–300 CrossRef CAS.
- F. L. Lu, Y. Y. Wang, C. Jin, F. Li, R. Z. Yang and F. L. Chen, J. Power Sources, 2015, 293, 726–733 CrossRef CAS.
- N. Sun, H. X. Liu, Z. Y. Yu, Z. N. Zheng and C. Y. Shao, Solid State Ionics, 2014, 268, 125–130 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- D. Thiele and A. Zuttel, J. Power Sources, 2008, 183, 590–594 CrossRef CAS.
- L. X. Wang, M. Ara, K. Wadumesthrige, S. Salley and K. Y. Simon, J. Power Sources, 2013, 234, 8–15 CrossRef CAS.
- G. Q. Zhang, J. P. Zheng, R. Liang, C. Zhang, B. Wang, M. Au, M. Hendrickson and E. J. Plichta, J. Electrochem. Soc., 2011, 158, A822–A827 CrossRef CAS.
- Y. Gao, C. Wang, W. Pu, Z. Liu, C. Deng, P. Zhang and Z. Mao, Int. J. Hydrogen Energy, 2012, 37, 12725–12730 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |