
A novel functional group difference-based selective etching strategy for the synthesis of hollow organic silica nanospheres†
Fanlong Zenga,
Lianxi Chen*a,
Jie Liab,
Xinshan Yec,
Huogen Yu a and
Zhenhui Liua
a and
Zhenhui Liua
aSchool of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, Wuhan, 430070, P. R. China. E-mail: clx@whut.edu.cn
bSchool of Materials Science and Engineering, Wuhan University of Technology, Wuhan, 430070, P. R. China
cStake Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing, 100191, P. R. China
First published on 8th March 2016
Abstract
A facile and effective “functional group difference-based selective etching” strategy has been developed to prepare organic functionalized hollow silica nanospheres (OHSNSs) with well-defined morphology and uniform size. The key point of the strategy is to introduce different organic groups into both core and shell for the purpose of changing their relative stability against etching, which results in the preferential etching of the organic functionalized inner core. In this paper, bifunctionalized core–shell silica nanospheres synthesized by using 2-cyanoethyltriethoxysilane (CTES) as the core and 3-thiocyanatopropyltriethoxysilane (TCPTES) as the shell can be easily transformed to thiocyanato group-functionalized hollow silica nanospheres (TC-HSNSs) in a Na2CO3 solution, based on the stability difference between the cyano group functionalized inner core and the thiocyanato group functionalized outer shell. Transmission electron microscopy (TEM) confirms that the formation of TC-HSNSs undergoes the process of selectively etching the inner core. Moreover, Fourier transform infrared (FTIR), energy dispersive spectroscopy (EDS) and elemental analysis (EA) prove that only the thiocyanato group is observed in the final product. In addition, the application of TC-HSNSs in the adsorption of aspirin has also been investigated.
1 Introduction
Hollow silica nanospheres (HSNSs) have attracted much research interest due to their great potential application in many fields, such as drug delivery, catalysis, sensors, and environmental protection.1–7 A series of synthetic methods have been developed to prepare HSNSs. To date, the most typical preparation methods for HSNSs are the templating methods, including hard-templating and soft-templating methods.8–14 However, for the hard-template methods, the preparation process is usually leads to hollow structure collapse during removal of sacrificial templates. Meanwhile, when the soft templating methods are adopted, the morphology and size distribution of HSNSs are hard to be controlled precisely.15Recently, researchers have explored a so-called self-templating route to prepare HSNSs in order to overcome the above-mentioned disadvantages.16–23 Shi's groups16 proposed a “structural difference-based selective etching” route to fabricate HSNSs. The process of HSNSs preparation involved fabrication of pure silica core/mesoporous silica shell nanoparticles and subsequent selectively removing pure silica core with appropriate etching agent based on structure difference between pure silica core and mesoporous silica shell. Zheng et al.17 developed a “cationic surfactant assisted selective etching” strategy to synthesize HSNSs. They found that the formation of the CTAB/SiO2 hybrid shell could effectively resist chemical etching, and accordingly inner pure silica core was preferentially etched. Very recently, self-templating methods had been developed for synthesizing OHSNSs.24–30 Shi and co-workers24 reported a “in situ hydrophobic layer protected selective etching” method to fabricate amino group functionalized HSNSs, in which the introduced amino group could form hydrophobic protecting layers on the surface of pure silica core/organic hybrid silica shell nanospheres, and consequently the relatively unstable pure silica core was selectively dissolved. In above-mentioned self-templating methods, the key point was that hybrid silica shell was more stable against etching than pure silica inner core. However, to our knowledge, some of hybrid silicas are more chemically inert than the pure silica,31 and thus OHSNSs are difficult to be obtained effectively. Therefore, pursuing appropriate synthetic methods to various OHSNSs are extremely desired.
It is well known that the solid-to-hollow transformation of the core–shell silica nanospheres is attributed to stability difference between core and shell in etching process. Herein, we present a novel “functional group difference-based selective etching” route to prepare OHSNSs with well-defined morphology and uniform size. The key point is that both core and shell are introduced different organic functional groups. When the stabilities of the core and shell are distinct different, the OHSNSs can be easily prepared. Compared with current self-templating methods, our strategy has three obvious advantages. Firstly, the preparation of TC-HSNSs does not require any assistant agents. Secondly, TC-HSNSs can be fabricated without the need for post-modification. Thirdly, it may be widely applied to fabricate various OHSNSs. In this paper, simply by treating the bifunctionalized core–shell silica nanospheres, which are synthesized by using CTES and TCPTES as precursors under hydrothermal condition, with a Na2CO3 solution, TC-HSNSs are easily synthesized based on stability difference between the cyano group functionalized inner core and the thiocyanato group functionalized outer shell. Additionally, the application of the TC-HSNSs in drug loading is also researched using aspirin as model drug.
2 Experimental section
2.1 Materials
Tetraethyl orthosilicate (TEOS, 96%), 3-thiocyanatopropyltriethoxysilane (TCPTES, 97%), ammonia hydroxide (25–28%), sodium carbonate, absolute ethanol, aspirin (98%) were purchased from Sinopharm Chemical Reagent Co. Cyanoethyltriethoxysilane (CTES, 95%), vinyltriethoxysilane (VTES, 99%) were obtained from Aladdin Chemistry Co. Ltd. Deionized water was used for all syntheses.2.2 Synthesis of CTES@TCPTES and VTES@TCPTES
Firstly, CTES and TCPTES were chosen as an example to prepare CTES@TCPTES. In a typical process, 30 mL of water and 1 mL of CTES were mixed with each other and stirred for 10 min in a round-bottom flask. After the addition of 1 mL of ammonia hydroxide solution, the mixture was further stirred at room temperature for 3 h. Then, 0.5 mL of TCPTES was added into the above suspension and stirred at room temperature for 5 h. Later, CTES@TCPTES could be obtained by two ways: hydrothermal treatment and non-hydrothermal treatment. For the hydrothermal treatment, the suspension was transferred into a sealed Teflon-lined autoclave and heated at 85 °C for 15 h. For non-hydrothermal treatment, the mixture was continuously stirred at room temperature for 15 h. Finally, suspension of CTES@TCPTES nanoparticles obtained by hydrothermal treatment or non-hydrothermal treatment was centrifuged and washed with ethanol for three times, and subsequently the product was dried at 50 °C for 12 h. For the preparation of VTES@TCPTES from VTES and TCPTES, the procedures and conditions were the same as those of corresponding CTES@TCPTES from CTES and TCPTES.2.3 Synthesis of TEOS@TCPTES spheres
3 mL TEOS was rapidly added into the mixture of 40 mL of ethanol, 2 mL of H2O, and 2 mL of ammonia hydroxide solution. Then the mixture was stirred at room temperature for 4 h, forming a white silica colloidal suspension. The solid SiO2 (sSiO2) particles were then obtained by centrifugation and washed with water three times. Later, 50 mg of the as-synthesized sSiO2 was dispersed in deionized water by ultrasonication for 10 min. The suspension was then added into a solution that consists of deionized water (15 mL), ethanol (15 mL), and ammonia hydroxide (0.3 mL). After the mixture was stirred at room temperature for 10 min, 0.2 mL of TCPTES was added quickly. Subsequently, the suspension was reacted at room temperature for 5 h, and then transferred into a sealed Teflon-lined autoclave and heated at 85 °C for 15 h. Ultimately, TEOS@TCPTES was then obtained by centrifugation, washed with ethanol three times, and dried at 50 °C for 12 h.2.4 Synthesis of TC-HSNSs
50 mg CTES@TCPTES was dispersed in 10 mL deionized water by ultrasonication for 10 min, then the suspension was added to 10 mL of Na2CO3 (0.2 g) aqueous solution. After the mixture was stirred at 50 °C for 10 h, the product was then obtained by centrifugation, washed ethanol for three times, and dried at 50 °C for 12 h. The same process as above was used to prepare TC-HSNSs from VTES@TCPTES and TEOS@TCPTES, respectively.2.5 Adsorption of aspirin for TC-HSNSs, TC-sSiO2, sSiO2

2.6 Characterization
Transmission electron microscopy (TEM) and energy dispersive spectroscopy (EDS) were conducted with a JEM 2100 F electron microscope operated at 200 kV. Scanning electron microscopy (SEM) images were determined by JSM-5610LV microscope. Fourier transform infrared (FTIR) spectra were collected with a Nicolet Nexus 470 IR spectrometer with KBr pellet. Nitrogen adsorption–desorption isotherms were obtained on Micromeritics ASAP 2020 system. Elemental analysis (EA) was measured with a Vario EL cube ANALYZER (CHNSO). UVvis spectra were recorded on a PerkinElmer Lambda 750S spectrophotometer.3 Results and discussion
In this paper, the synthesis of TC-HSNSs was schematically illustrated in Scheme 1. In a typical process, monodispersed solid silica spheres were firstly prepared by the hydrolysis of CTES under alkaline conditions using ammonia as the catalyst, and then a hybrid silica shell was formed on the surface of each aforementioned solid silica sphere by the hydrolysis and co-condensation of TCPTES, forming solid core/shell structures, denoted as CTES@TCPTES. Finally, the solid silica cores were etched away completely or partially by using a mild etching agent, Na2CO3. In contrast experiment, VTES@TCPTES and TEOS@TCPTES were also prepared, and then etched in a Na2CO3 solution.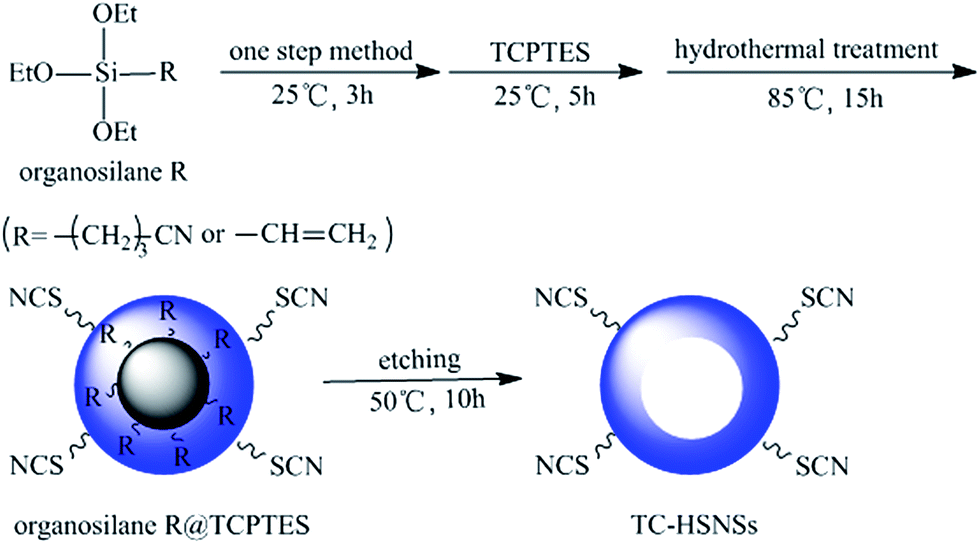 | ||
| Scheme 1 Schematic illustration for the synthesis and structure evolution of TC-HSNSs. | ||
3.1 Synthesis of TC-HSNSs from bifunctionalized core–shell silica nanospheres
Fig. 1a–c showed TEM images of CTES@TCPTES by hydrothermal treatment at different etching time. When the nanospheres were etched for 5 h, there was obvious contrast between the C-sSiO2 core and TC-sSiO2 shell (Fig. 1a). The C-sSiO2 core was gradually dissolved while the TC-sSiO2 shell remained intact. While the etching time was extended to 10 h, the solid bifunctionalized core–shell nanospheres were completely converted into TC-HSNSs as demonstrated by typical images by TEM (Fig. 1b). SEM images (Fig. 3e) showed that the as-obtained TC-HSNSs had a high monodispersity and uniform size. Meanwhile, the surface of TC-HSNSs became rough, proving the existence of pores. The BET surface area and average pore size of TC-HSNSs were 47.5 m2 g−1 (Fig. 2a) and 6.6 nm (Fig. 2b), respectively. In comparison, the BET surface area and average pore size of CTES@TCPTES were only 2.9 m2 g−1 (Fig. 2a) and 0.8 nm (Fig. 2b), respectively. However, as the etching time was prolonged to 15 h, it was clearly found that part of shell of TC-HSNSs had been damaged (Fig. 1c). | ||
| Fig. 1 TEM images of CTES@TCPTES by hydrothermal treatment in Na2CO3 solution at 50 °C for (a) 5 h and (b) 10 h and (c) 15 h, respectively; VTES@TCPTES by hydrothermal treatment in Na2CO3 solution at 50 °C for (d) 5 h and (e) 10 h and (f) 15 h, respectively; TEOS@TCPTES by hydrothermal treatment in Na2CO3 solution at 50 °C for (g) 5 h and (h) 10 h and (i) 15 h, respectively. | ||
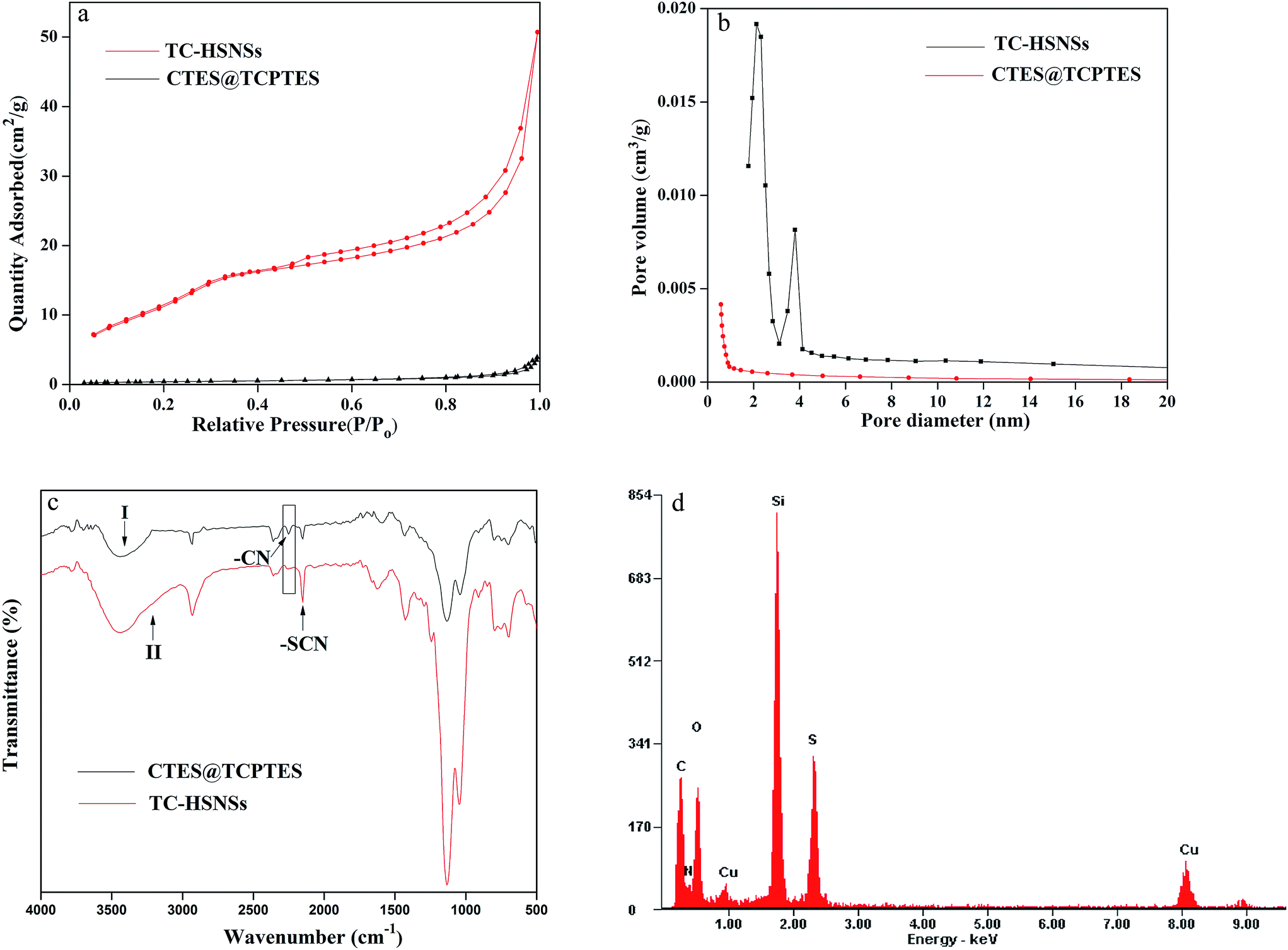 | ||
| Fig. 2 N2 adsorption–desorption isotherms (a) and pore size distributions (b) of typical TC-HSNSs and CTES@TCPTES, respectively. (c) FTIR spectra of CTES@TCPTES (curve I) and TC-HSNSs made from CTES@TCPTES (curve II), respectively. (d) EDS spectrum of TC-HSNSs made from CTES@TCPTES. | ||
Another control experiment was also performed with VTES@TCPTES by hydrothermal treatment. Fig. 1d–f represented the process in which the core–shell structure was converted into the hollow structure. In the first 5 h, solid microspheres showed little change (Fig. 1d). Until the etching time was prolonged to 10 h, the vinyl functionalized solid silica (V-sSiO2) core was gradually dissolved, and a few of the hollow spheres appeared (Fig. 1e). When the etching time was further extended to 15 h, hollow structure was more obvious (Fig. 1f). Compared to the solid VTES@TCPTES, the hollow nanospheres maintained their original morphology, smoothness, and initial size, implying that the inner structure was preferentially etched in Na2CO3 solution.
In addition, TEOS@TCPTES colloids by hydrothermal treatment were also treated using a Na2CO3 solution. Surprisingly, when TEOS@TCPTES was etched at different stages, no obvious etching of the sSiO2 core was observed from Fig. 1g–i. In addition, the surface of TEOS@TCPTES was almost smooth, and distinct size reduction of TEOS@TCPTES had been not discovered, and these results proved that solid SiO2 core was not etched.
The obtained TC-HSNSs from CTES@TCPTES were further characterized by Fourier transform infrared (FTIR), energy dispersive spectroscopy (EDS) and elemental analysis (EA). In the FTIR spectrum of CTES@TCPTES by hydrothermal treatment (Fig. 2c, curve I), the stretching vibration peaks at 2251 cm−1 and 2152 cm−1 could be attributed to cyano group and the thiocyanato group, respectively.33,34 Whereas stretching vibration band of cyano group was almost not observed in FTIR spectrum of TC-HSNSs (Fig. 2c, curve II). The results showed that C-sSiO2 core might be completely etched and thiocyanato group in TC-HSNSs was retained.
In order to provide more evidences to explain the variation before and after etching, the elemental distribution of the TC-HSNSs and element content ratio of both CTES@TCPTES by hydrothermal treatment and TC-HSNSs were recorded by EDS spectrum and EA, respectively. In Fig. 2d, the EDS spectrum of TC-HSNSs demonstrated the presence of six elements including carbon, nitrogen, oxygen, silicon, sulfur and copper, in which the copper came from copper wire mesh that was used to hold samples. Table 1 showed that value of N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S of TC-HSNSs was higher than that of CTES@TCPTES. Because CTES@TCPTES contained two organic groups: –CN and –SCN, it leaded to high nitrogen content and low sulfur content. However, TC-HSNSs only contained –SCN, so the value of N:S was lower. These results demonstrated that the original CTES@TCPTES was successfully turned into TC-HSNSs.
:S of TC-HSNSs was higher than that of CTES@TCPTES. Because CTES@TCPTES contained two organic groups: –CN and –SCN, it leaded to high nitrogen content and low sulfur content. However, TC-HSNSs only contained –SCN, so the value of N:S was lower. These results demonstrated that the original CTES@TCPTES was successfully turned into TC-HSNSs.
| Sample | N (%) | S (%) |
|---|---|---|
| CTES@TCPTES | 11.54 | 8.26 |
| TC-HSNSs | 9.45 | 18.23 |
3.2 Synthesis of TC-HSNSs from CTES@TCPTES in different etching temperature
To reveal the influence of etching temperature in the synthesis of TC-HSNSs, two groups of control experiments were implemented. Fig. 3a and b showed TEM images of CETS@TCPTES by hydrothermal treatment in Na2CO3 solution for 10 h at 35 °C and 65 °C, respectively. In Fig. 3a, the samples were only partially etched and slightly aggregated. However, from Fig. 3b, it could be observed that the solid-to-hollow conversion was accomplished and a large number of TC-HSNSs were aggregated and damaged. It was demonstrated that the rate of solid-to-hollow structure conversion could be tuned by changing the reaction temperature. While the reaction temperature was too high (for instance 65 °C), the dispersity of prepared TC-HSNSs would lower. Therefore, an appropriate etching temperature (for instance 50 °C) was thus indispensable for preparation of high-quality product. | ||
| Fig. 3 TEM images of CTES@TCPTES by hydrothermal treatment in Na2CO3 solution at (a) 35 °C for 10 h and (b) 65 °C for 10 h, SEM images of the initial CTES@TCPTES by hydrothermal treatment (c) and non-hydrothermal treatment (d), respectively, TC-HSNSs made from CTES@TCPTES by hydrothermal treatment (e) and non-hydrothermal treatment (f) in Na2CO3 solution at 50 °C for 10 h, respectively, TEM images of CTES@TCPTES (g) by non-hydrothermal treatment in Na2CO3 solution at 50 °C for 10 h. | ||
3.3 Synthesis of TC-HSNSs from CTES@TCPTES by non-hydrothermal treatment
In order to explore the influence of hydrothermal treatment in the formation of CTES@TCPTES, CTES@TCPTES was prepared by non-hydrothermal treatment. Fig. 3c and d showed that the morphology and size distribution of CTES@TCPTES by hydrothermal treatment and non-hydrothermal treatment, respectively. Compared with the results displayed in Fig. 3c, the original CTES@TCPTES had a wide size distribution (500–1200 nm). It was indicated that hydrothermal treatment was contributed to form CTES@TCPTES with uniform size (460 nm). To research influence of hydrothermal process for forming TC-HSNSs, the original CTES@TCPTES was etched with Na2CO3. Notably, when the original sample was completely turned into the TC-HSNSs after etching, the product with large size was disappeared and most of the TC-HSNSs with small size had shell collapsed and deformed (Fig. 3g). The morphology of nanospheres after etching was illustrated by SEM images (Fig. 3f), and it showed that the massive TC-HSNSs had appeared aggregation and breakage. Nevertheless, as illustrated in Fig. 3e, the obtained TC-HSNSs from CTES@TCPTES by hydrothermal treatment had maintained regular spheres and highly monodisperse. The results revealed that structure of CTES@TCPTES by hydrothermal treatment was more stable than that of CTES@TCPTES by non-hydrothermal treatment. Therefore, hydrothermal treatment was critical for preparation of high-quality TC-HSNSs. In the conversion process of CTES@TCPTES to TC-HSNSs, hydrothermal treatment should at least play the following two important roles: (1) promoted fabrication of CTES@TCPTES with uniform size; (2) facilitated structural stability of CTES@TCPTES in Na2CO3 solution.3.4 Adsorption of aspirin
Drug adsorption property of the TC-HSNSs was evaluated by using aspirin as a model molecule. Meanwhile, sSiO2 and TC-sSiO2 were also used for comparison. As shown in Fig. 4, the adsorption process of the curve b and curve c was similar, and it involved the rapid adsorption of aspirin within the first 8 h, and then reached approximate equilibrium with only small changes from 8–16 h. It is believed that the adsorption of aspirin on the surface of sSiO2 and TC-sSiO2 was mainly ascribed to hydrogen bonding interaction between aspirin molecules and functional groups of nanospheres surface. Nevertheless, the amounts of aspirin adsorption for sSiO2 were slightly less than that for TC-sSiO2 at the same time. One possible reason was that aspirin molecules prefered to bond functional groups of the surface of TC-sSiO2 instead of that of sSiO2. When the adsorption time was above 16 h, the amounts of aspirin adsorption were slightly decreased to 6.3% (curve b) and 5.1% (curve c), respectively. It was speculated that the adsorption of aspirin on the surface of sSiO2 and TC-sSiO2 because of the process of physical absorption could be released from the surface of nanoparticles. Compared with curve b, c, curve a showed a process of rapid adsorption of aspirin. In initial 12 h, the adsorption efficiency of aspirin for TC-HSNSs reached at 13.5%, which was far more than the above two samples. Whereafter, the adsorption efficiency of aspirin tended to balance (14.7%) from 12–20 h. The adsorbing efficiency of aspirin was slightly declined at last 4 h. From Fig. 4, it could be discovered that the adsorbing efficiency of three samples for aspirin decreased as TC-HSNSs > TC-sSiO2 > sSiO2. According to the experiment results, the high loaded aspirin for TC-HSNSs could be attributed to thiocyanato groups and the hollow structure of TC-HSNSs. | ||
| Fig. 4 Adsorption of aspirin for (a) TC-HSNSs and (b) TC-sSiO2 and (c) sSiO2, respectively. | ||
4 Conclusions
In summary, a novel and general functional group difference-based selective etching strategy has been developed to fabricate OHSNSs with well-defined morphology and uniform size. The key in this strategy is to introduce different organic functional group into both core and shell so that their relative stability is changed. From the synthesis process of TC-HSNSs, two facts are discovered. Firstly, organic silica core is more favourite for the formation of TC-HSNSs relative to pure silica core. Secondly, the greater the stability difference between core and shell is, the easier the TC-HSNSs are obtained. Moreover, the morphology and size distribution of the products can be influenced by etching temperature and hydrothermal process. If an appropriate etching temperature and hydrothermal process are adopted, TC-HSNSs with well-defined morphology and uniform size distribution can be easily fabricated. Although this study mainly focuses on the preparation of TC-HSNSs, it is expected that the preparation method may be extended applied to fabricate other OHSNSs. As long as there is adequate stability different between inner core and outer shell, the OHSNSs can be effectively prepared. As a simple application, the TC-HSNSs have been used as vehicle for aspirin adsorption and show high loading amounts for aspirin.Acknowledgements
This work is financially supported by the Nature Science Foundation of Hubei Province (No. 2014CFB862), the State Key Laboratory of Natural and Biomimetic Drugs (No. K20140214) and the Fundamental Research Funds for the Central Universities (WUT: 2015IB002). We also would like to thank Xiao Qing Liu and Ting Ting Luo for their HRTEM analysis in the Materials Research and Test Center of WUT.References
- Y. S. Li and J. L. Shi, Adv. Mater., 2014, 26, 3176–3205 CrossRef CAS PubMed
.
- X. W. Lou, A. A. Lynden and Z. C. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS
- Y. F. Zhu, J. L. Shi, Y. S. Li, H. R. Chen, W. H. Shen and X. P. Dong, Microporous Mesoporous Mater., 2005, 85, 75–81 CrossRef CAS
- X. Q. Huang, H. H. Zhang, C. Y. Guo, Z. Y. Zhou and N. F. Zheng, Angew. Chem., 2009, 48, 4808–4812 CrossRef CAS PubMed
- M. Retsch, M. Schmelzeisen, H. J. Butt and E. L. Thomas, Nano Lett., 2011, 11, 1389–1394 CrossRef CAS PubMed
- S. W. Liu, J. G. Yu and J. Mietek, J. Am. Chem. Soc., 2010, 132, 11914–11916 CrossRef CAS PubMed
- Z. G. Teng, X. D. Su, Y. Y. Zheng, J. Sun, G. T. Chen, C. C. Tian, J. D. Wang, H. Li, Y. N. Zhao and G. M. Lu, Chem. Mater., 2013, 25, 98–105 CrossRef CAS
- G. G. Qi, Y. B. Wang, L. Estevez, A. K. Switzer, X. N. Duan, X. F. Yang and E. P. Giannelis, Chem. Mater., 2010, 22, 2693–2695 CrossRef CAS
- Z. Chen, Z. M. Cui, F. Niu, L. Jiang and W. G. Song, Chem. Commun., 2010, 46, 6524–6526 RSC
- Z. W. Deng, M. Chen, S. X. Zhou, B. You and L. M. Wu, Langmuir, 2006, 22, 6403–6407 CrossRef CAS PubMed
- H. J. Zhang, J. Wu, L. P. Zhou, D. Y. Zhang and L. M. Qi, Langmuir, 2007, 23, 1107–1113 CrossRef CAS PubMed
- Y. L. Chen, Y. Li, Y. X. Chen, X. J. Liu, M. Zhang, B. Z. Li and Y. G. Yang, Chem. Commun., 2009, 5177–5179 RSC
- J. G. Wang, F. Li, H. J. Zhou, P. C. Sun, D. T. Ding and T. H. Chen, Chem. Mater., 2009, 21, 612–620 CrossRef CAS
- M. Chen, L. M. Wu, S. X. Zhou and B. You, Adv. Mater., 2006, 18, 801–806 CrossRef CAS
- Q. Zhang, W. S. Wang, J. Goebl and Y. D. Yin, Nano Today, 2009, 4, 494–507 CrossRef CAS
- Y. Chen, H. R. Chen, L. M. Guo, Q. J. He, F. Chen, J. Zhou, J. W. Feng and J. L. Shi, ACS Nano, 2010, 1, 529–539 CrossRef PubMed
- X. L. Fang, C. Chen, Z. H. Liu, P. X. Liu and N. F. Zheng, Nanoscale, 2011, 3, 1632–1639 RSC
- T. R. Zhang, Q. Zhang, J. P. Ge, G. James, M. W. Sun, Y. S. Yan, Y. S. Liu, C. L. Chang, J. H. Guo and Y. D. Yin, J. Phys. Chem. C, 2009, 113, 3168–3175 CAS
- Q. Zhang, J. P. Ge, G. James, Y. X. Hu, Z. D. Lu and Y. D. Yin, Nano Res., 2009, 2, 583–591 CrossRef CAS
- D. Chen, L. L. Li, F. Q. Tang and S. Qi, Adv. Mater., 2009, 21, 3804–3807 CrossRef CAS
- Q. Y. Yu, P. P. Wang, S. Hu, J. F. Hui, J. Zhuang and X. Wang, Langmuir, 2011, 27, 7185–7191 CrossRef CAS PubMed
- Q. Y. Yu, J. F. Hui, P. P. Wang and X. Wang, Inorg. Chem., 2012, 51, 9539–9543 CrossRef CAS PubMed
- Y. Yang, J. Liu, X. B. Li, X. Liu and Q. H. Yang, Chem. Mater., 2011, 23, 3676–3684 CrossRef CAS
- K. Zhang, H. R. Chen, Y. Y. Zheng, Y. Chen, M. Ma, X. Wang, L. J. Wang, D. P. Zeng and J. L. Shi, J. Mater. Chem., 2012, 22, 12553–12561 RSC
- S. Shi, M. Wang, C. Chen, F. Lu, X. Zheng, J. Gaoa and J. Xu, RSC Adv., 2013, 3, 1158–1164 RSC
- N. Koike, T. Ikuno, T. Okubo and A. Shimojima, Chem. Commun., 2013, 49, 4998–5000 RSC
- Y. Chen, Q. S. Meng, M. Y. Wu, S. G. Wang, P. F. Xu, H. R. Chen, Y. P. Li, L. X. Zhang, L. Z. Wang and J. L. Shi, J. Am. Chem. Soc., 2014, 136, 16326–16334 CrossRef CAS PubMed
- Z. G. Teng, X. D. Su, B. H. Lee, C. G. Huang, Y. Liu, S. J. Wang, J. Wu, P. Xu, J. Sun, D. K. Shen, W. Li and G. M. Lu, Chem. Mater., 2014, 26, 5980–5987 CrossRef CAS
- J. W. Park, J. S. Kim, T. J. Park, E. H. Kim and S. M. Koo, J. Colloid Interface Sci., 2015, 438, 220–226 CrossRef CAS PubMed
- M. Y. Wu, Y. Chen, L. X. Zhang, X. Y. Li, X. J. Cai, Y. Y. Du, L. L. Zhang and J. L. Shi, J. Mater. Chem. B, 2015, 3, 766–775 RSC
- J. Li, L. X. Chen, X. Li, C. C. Zhang and F. L. Zeng, Appl. Surf. Sci., 2015, 340, 126–131 CrossRef CAS
- J. L. Pang, X. Y. Li, G. W. Zhou, B. Suna and Y. Q. Wei, RSC Adv., 2015, 5, 6599–6606 RSC
- F. P. Dong, W. P. Guo, S. K. Park and C. S. Ha, Chem. Commun., 2012, 48, 1108–1110 RSC
- Z. C. Li, H. T. Fanb, Y. Zhang, M. X. Chena, Z. Y. Yua, X. Q. Cao and T. Sun, Chem. Eng. J., 2011, 171, 703–710 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra25161c |
| This journal is © The Royal Society of Chemistry 2016 |