
A salification-induced charge transfer effect for improving the resistive memory performance of azo derivative-based devices†
Quan Liua,
Qingfeng Xua,
Huilong Dongb,
Hua Lia,
Dongyun Chena,
Lihua Wanga,
Youyong Lib and
Jianmei Lu*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Key Laboratory of Adsorption Technology in Petroleum and Chemical Industry for Wastewater Treatments, Soochow University, 199 Ren'ai Road, Suzhou 215123, China. E-mail: lujm@suda.edu.cn; Fax: +86 512 65880367; Tel: +86 512 65880368
bFunctional Nano & Soft Materials Laboratory (FUNSOM) and Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, Suzhou, 215123, China
First published on 19th January 2016
Abstract
In this study, we report the synthesis of a new organic conjugate molecule, 3-(4-((4-(dimethylamino)phenyl)diazenyl)phenyl)-1-(pyridin-4-yl)prop-2-en-1-one (AZOCP), and its camphorsulfonic acid salt (AZOCP–CSA). The photophysical and electrochemical characterization reveals that an enhanced π–π conjugation is formed in the camphorsulfonic acid salt because of the salification effect. The salification reaction also plays an important role in the formation of a more ordered stacking nanocrystalline film as evidenced by AFM and XRD analysis, and thus gives rise to an improved transport of charge carriers. The comparison of device performance demonstrates that the device based on the use of the salificated compound has better resistive memory behaviour in terms of ON/OFF ratio, retention time and rewritable cycle. Isothermal I–V correction and theoretical calculations confirm that the resistive performance is a result of an electric-field-induced charge transfer effect and the enhanced device performance of camphorsulfonic acid salt is due to the presence of a strong salification-induced charge transfer effect. Our experimental findings suggest that the simple but effective salification strategy may find widespread use in promoting performance of other organic resistive memory devices by introducing a strong charge transfer effect.
Introduction
Since resistive behavior was proposed, a great deal of research interest has been devoted to the development of various resistive materials for practical application.1–4 The resistive memory is recognized as an appealing candidate for the next generation “universal memory” due to its high density, low power consumption, large ON/OFF ratio, high endurance and long retention.5–10 Therefore, a variety of materials and strategies for memory application has been provided.11,12 Among them, organic small molecule based memories due to their tunable functionalization and easy device miniaturization are becoming the most promising materials to realize high-performance resistive performance.13–16A variety of design approaches for organic molecule-based resistive memory devices have already been reported in the literature.17–28 To tune the memory performance, one of the most common methods is blending with other salt or dyes.29 for instance, Meskers once introduced a series of inorganic salt (LiCF3SO3 or NaCl) in the polymer layer results in resistive switching behavior under external electric field.30,31 It has been suggested that the migration of the dopant ions into and out of the polymer depletion layer at the aluminum Schottky contact causes resistive bistability behavior. Our group previously reported facilely blending the donor and acceptor moieties of the D–π–A framework as the active materials for resistive switching memory, and the change of the blend ratio to tune electrical data storage performance from volatile to non-volatile.32 However, the proportion of the blend part has great effect on the performance, and the inevitable separation of the blend phase will also have a great impact on the memory performance.
The method of salification not only can effectively promote the thermal stability of the compound,33 but also can improve the uniformity and stability of the films.34 Li et al. has designed and reported protonic-acid-doped poly(Schiff base) resistive memory devices, which has long consecutive switching cycles and excellent operative uniformity.35 However, the application of salification to tune the memory performance of small molecules was seldom reported.
In this work, we synthesized a new conjugated molecule containing a pyridine as end group: 3-(4-((4-(dimethylamino)phenyl)diazenyl)phenyl)-1-(pyridin-4-yl)prop-2-en-1-one (AZOCP). Then, camphorsulfonic acid (CSA) was used as a salification agent to formed AZOCP–CSA.36 The AZOCP–CSA can be effectively obtained by tuning the AZOCP/CSA ratio, which can be accurately confirmed by 1H NMR spectroscopy (Fig. S4†). However, when increasing the amount of camphorsulfonic acid to change the AZOCP/CSA ratio to 1/2, the compound can not avoid moisture absorption in the air and hard to forming a film. By contrast, AZOCP–CSA with ratio of 1/1 has good solubility, thermal stability and fine film-forming properties. Then, fabricated Au/AZOCP/ITO and Au/AZOCP–CSA/ITO devices and their resistance-switching parameters have been studied in details. Specifically, the Au/AZOCP–CSA/ITO device shows a greater performance improvement and a promising potential for information storage applications.
Experimental
Materials
Sodium nitrite, 4-aminobenzaldehyde polymer, anhydrous sodium acetate, N,N-dimethylaniline, 1-(pyridin-4-yl)ethanone, camphorsulfonic acid were purchased from commercial sources (TCI, Alfa Aesar, and Sigma-Aldrich). All solvents were purchased from Sinopharm Chemical Reagent Co., Ltd. All chemicals were used as received without further purification.Characterization
All electrical measurements of the device were characterized under ambient conditions without any encapsulation using Keithley 4200 semiconductor characterization system in voltage sweeping mode. The sweeping step is 0.01 V. 1H nuclear magnetic resonance (1H NMR) spectra were measured at 400 MHz on a Bruker 400 AVANCE spectrometer with dimethyl sulfoxide-d6 (DMSO-d6) as the solvent. High-resolution mass spectra (HRMS) were acquired using a Micro mass GCT-TOF mass spectrometer with an electrospray ionization source. UV-vis absorption spectra were recorded on a Shimadzu UV-3600 spectrophotometer at room temperature. Thermogravimetric analysis (TGA) was performed on a Perkin-Elmer Diamond TG/DTA instrument under a nitrogen atmosphere with the gas-flow rate of 50 mL min−1 and a heating rate of 10 °C min−1. Cyclic voltammetry (CV) measurements were carried out under an argon atmosphere. The film coated on a ITO electrode (working electrode) was scanned at 1 mV s−1 in a 0.1 M solution of tetrabutylammonium hexafluorophosphate (n-Bu4NPF6) in acetonitrile, with Ag/AgCl (3.8 M KCl) and a platinum as the reference and counter electrodes, respectively. The atomic force microscopy (AFM) measurements were performed using a MFP-3DTM (Digital Instruments/Asylum Research) AFM instrument in the tapping mode. X-ray diffraction (XRD) patterns were collected using an X'Pert-Pro MPD X-ray diffractometer.Fabrication of memory devices
The ITO substrates were pre-cleaned with ethanol, acetone and isopropanol sequentially in an ultrasonic bath for 20 min. A 0.1 mL benzene solution of AZOCP or AZOCP–CSA was spin-coated onto the substrates at a spinning speed of 500 rpm for 10 s and then 2000 rpm for 30 s, followed by a vacuum-drying at 80 °C for 8 h. Before spin-coating, the solution was filtered through polytetrafluoroethylene (PTFE) membrane micro-filters with a pore size of 0.32 μm. The thickness of the films is about 90 nm as measured by spectroscopic ellipsometer (model M2000DI, Woollam). To construct the Au/AZOCP or AZOCP–CSA/ITO structures, Au top electrodes was thermally evaporated onto the film surface under 2 × 10−6 Torr through a shadow mask with thickness around 60 nm and area of 0.20 mm2.Synthetic procedures
A solution of sodium nitrite (4.35 g, 0.063 mol) in water (24 mL) was added dropwise to a mixture of 4-aminobenzaldehyde polymer (6.03 g, 0.06 mol), water (32 mL) and concentrated hydrochloric acid (20 mL) at 0–5 °C. The mixture was stirred at 0–5 °C for 30 min. A mixture of N,N-dimethylaniline (8.0 g, 0.066 mol), concentrated hydrochloric acid (10 mL) and water (30 mL) was added slowly to the diazonium salt solution at 0–5 °C. After 1 h, anhydrous sodium acetate (18 g, 0.22 mol) was added to the resulting mixture, which was then stirred at 0–5 °C for 24 h. The solution was filtered and the obtained crude product was recrystallized from ethanol to afford compound 1 as a brown powder (yield 75%).
1H NMR (400 MHz, DMSO-d6) d (ppm): 10.06 (s, 1H, CHO), 8.05 (d, J = 8.0 Hz, 2H, ArH), 7.93 (d, J = 8.0 Hz, 2H, ArH), 7.85 (d, J = 8.0 Hz, 2H, ArH), 6.86 (d, J = 8.0 Hz, 2H, ArH), 3.09 (s, 6H, 2CH3). 13C NMR (100 MHz, DMSO-d6): d (ppm): 192.4, 155.9, 153.1, 142.8, 136.0, 130.7, 125.5, 122.3, 111.6, 39.8.
1H NMR (400 MHz, CDCl3) d (ppm): 8.85 (d, J = 4.4 Hz, 2H, ArH), 7.92–7.86 (m, 5H, ArH),7.79 (d, J = 5.6 Hz, 2H, ArH), 7.76 (d, J = 8.4 Hz, 2H, ArH), 7.47 (d, J = 16 Hz, 1H, ArH), 6.77 (d, J = 9.2 Hz, 2H), 3.12 (s, 6H, 2CH3). 13C NMR (100 MHz, CDCl3): d (ppm): 189.2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 154.3 (Ar–C), 152.4 (Ar–C), 150.3 (Ar–C), 145.8 (Ar–C), 144.1 (Ar–C), 143.3 (Ar–C), 134.3 (Ar–C), 129.2 (Ar–C), 125.0 (Ar–C), 122.4 (Ar–C), 121.1 (Ar–C), 120.7 (Ar–C), 111.4 (CC), 39.8 (2CH3). HRMS: calcd for C22H20N4O [M + H]+ 357.1715, found 357.1667.
O), 154.3 (Ar–C), 152.4 (Ar–C), 150.3 (Ar–C), 145.8 (Ar–C), 144.1 (Ar–C), 143.3 (Ar–C), 134.3 (Ar–C), 129.2 (Ar–C), 125.0 (Ar–C), 122.4 (Ar–C), 121.1 (Ar–C), 120.7 (Ar–C), 111.4 (CC), 39.8 (2CH3). HRMS: calcd for C22H20N4O [M + H]+ 357.1715, found 357.1667.
Preparation of AZOCP–CSA
The protonation of AZOCP by camphorsulfonic acid (CSA) was performed in chloroform at room temperature. A CSA/chloroform solution was added to an AZOCP/chloroform solution with the CSA to AZOCP molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The salification process continued for one day to ensure thorough reaction. Chloroform was then removed by rotary evaporation followed by vacuum distillation at 80 °C for one day. By comparing the 1H NMR spectra of AZOCP–CSA in CDCl3, the ratio of the integral of CH3 in AZOCP and CH3 in CSA is 1:1 which means AZOCP and CSA are salified successfully (Fig. S4†).
:1. The salification process continued for one day to ensure thorough reaction. Chloroform was then removed by rotary evaporation followed by vacuum distillation at 80 °C for one day. By comparing the 1H NMR spectra of AZOCP–CSA in CDCl3, the ratio of the integral of CH3 in AZOCP and CH3 in CSA is 1:1 which means AZOCP and CSA are salified successfully (Fig. S4†).
Results and discussion
AZOCP and AZOCP–CSA can be obtained via the synthetic routes as shown in Scheme 1. The thermal stability of AZOCP and AZOCP–CSA were evaluated by TGA under a nitrogen atmosphere. As shown in the TGA curves (Fig. S5†), the thermal decomposition temperature (the 5% weight-lost temperature) of AZOCP was 194 °C and that of AZOCP–CSA was up to 213 °C. The good-thermal stability of AZOCP–CSA could better endure heat deterioration in the memory devices.38,39 | ||
| Scheme 1 The synthetic routes and molecular structures of AZOCP and AZOCP–CSA. | ||
Photophysical and electrochemical properties
In view of the AZOCP and AZOCP–CSA has good solubility in polar solvent. So we choose chlorobenzene as the solvent and used spin coated method to fabricate the film. Fig. 1 shows the optical absorption spectra of AZOCP and AZOCP–CSA nanofilms on quartz substrates. The weak energy absorption bands at 300–350 nm can be attributed to the π → π* transition of the azobenzene chromophore and the strong absorption bands at approximately 400–500 nm can be attributed to the n → π* transition (charge transfer) of the azobenzene.40,41 The optical band gaps of the AZOCP and AZOCP–CSA molecules, estimated from the absorption edges of the films, are 2.22 and 2.01 eV, respectively. Compared with that of AZOCP, the onset optical absorbance of AZOCP–CSA exhibits a significant red-shift for 21 nm, which corresponds to a narrower energy band gap of the salification system. Therefore, salificating AZOCP with camphorsulfonic acid gives rise to a ground state charge transfer complex in AZOCP–CSA. Meanwhile, it also suggests that the AZOCP–CSA film forms an ordered stacking of the π-conjugation system, favouring an effective carriers migration. | ||
| Fig. 1 UV-vis spectra of the AZOCP and AZOCP–CSA thin films on quartz substrates. | ||
Fig. 2 shows the cyclic voltammetry (CV) measurements of AZOCP and AZOCP–CSA on an indium-tin-oxide (ITO) glass substrate in a 0.1 mol L−1 solution of tetrabutylammonium hexafluorophosphate (TBAPF6) in anhydrous acetonitrile solution; measurements were taken with a scan rate of 100 mV s−1. The onset oxidation (Eonsetox) of AZOCP and AZOCP–CSA are approximately 0.88 V and 0.94 V vs. Ag/AgCl, respectively. The oxidation potential onset (Eonsetox) of ferrocene was measured to be 0.44 V vs. Ag/AgCl in acetonitrile with bare ITO glass. The estimated highest occupied molecular orbital (HOMO) levels can be calculated from the oxidation potential onset Eonsetox according to the following formula: HOMO = −[Eonsetox + 4.8 − EFc] eV. The lowest unoccupied molecular orbital (LUMO) levels are not detectable from CV; the values of the LUMO levels were estimated from following formula: LUMO = [HOMO + Eg] eV. The HOMO levels of AZOCP and AZOCP–CSA are −5.24 eV and −5.3 eV, respectively. The determined LUMO levels of AZOCP and AZOCP–CSA are −3.02 eV and −3.17 eV, respectively (see Table S1†). AZOCP–CSA has lower LUMO levels than AZOCP, which due to the charge transfer process, agreement with UV spectroscopic data.
 | ||
| Fig. 2 Cyclic voltammograms of AZOCP and AZOCP–CSA films on ITO substrates. | ||
Morphology of the thin film
To investigate the surface morphology and film microstructure, atomic force microscopy (AFM) and X-ray diffraction (XRD) measurements were carried out on the AZOCP and AZOCP–CSA films. As seen from the AFM height images (Fig. 3), both of molecule films showed a relatively smooth surface without obvious defects. The AZOCP film clearly showed two-dimensional grains in its solid state at room temperature with uniform size and the surface root-mean-square (RMS) roughness is 4.45 nm. The AZOCP–CSA film showed a denser needle morphology, and a RMS roughness of 2.5 nm, smaller than that of AZOCP. Therefore, salification improves the film quality significantly. | ||
| Fig. 3 Tapping-mode AFM topography (5 × 5 μm) of spin-coated AZOCP (a) and AZOCP–CSA (b) films on ITO substrates, respectively. | ||
In order to measure the crystalline order within the films, X-ray diffraction (XRD) experiments were performed on the spin-coated films. From the XRD patterns (Fig. 4), AZOCP films showed an unconspicuous diffraction peak at 2θ = 5.6° of AZOCP corresponds to a d-spacing of ca. 15.8 Å. However, the “counterpart” compound AZOCP–CSA film was observed a higher intensity peak at 2θ = 5.6° and another strong peak at 2θ = 11° and 21.6° are corresponding to the d-spacing distances of15.8 Å, 8.0 Å and 4.1 Å, respectively. Furthermore, one diffraction angle of AZOCP–CSA is approximately two times larger than the other. These results indicate that the AZOCP–CSA molecule formed a highly ordered crystalline and a close layer-by-layer stacking structure in the thin film state that is favorable for forming highly efficient pathways for charge carrier transport and to obtain good memory performance.42
 | ||
| Fig. 4 XRD patterns of AZOCP and AZOCP–CSA spin coated films onto quartz glass substrates. | ||
Current–voltage (I–V) characteristics of the memory devices
The memory effect of the AZOCP devices was explored first, and the current–voltage (I–V) characteristics are shown in Fig. 5a. The fabricated Au/AZOCP/ITO memory devices have a resistance of about 5.9 kohm, and are in high resistance state (HRS) or OFF state. The arrows indicate the voltage sweeping direction. Under a positively biased voltage sweeping, the current increases abruptly and reaches the compliance current (10−2 A) at around 2.5 V, indicating that the device has been switched from a HRS to a low resistance state (LRS or ON state) (sweep 1 of Fig. 5a). This transition can be defined as the “Write” or “SET” process and the ON/OFF ratio of ∼102 can be obtained. We defined ON/OFF ratio as RHRS/RLRS, where RHRS and RLRS are the resistance values in the HRS and LRS, respectively. The device remains at the ON state, even after the power is turned off or during the subsequent forward voltage sweeping (sweeps 2). Meanwhile, the current abruptly decreased during the sweep from 0 to −3 V, which is the transition from LRS to HRS. This transition serves as the “Erase” or “RESET” process of a rewritable device. The OFF state can be maintained in the final sweep from −3 to 0 V (sweeps 4). Therefore, AZOCP-based device has potential applications for non-volatile data storage.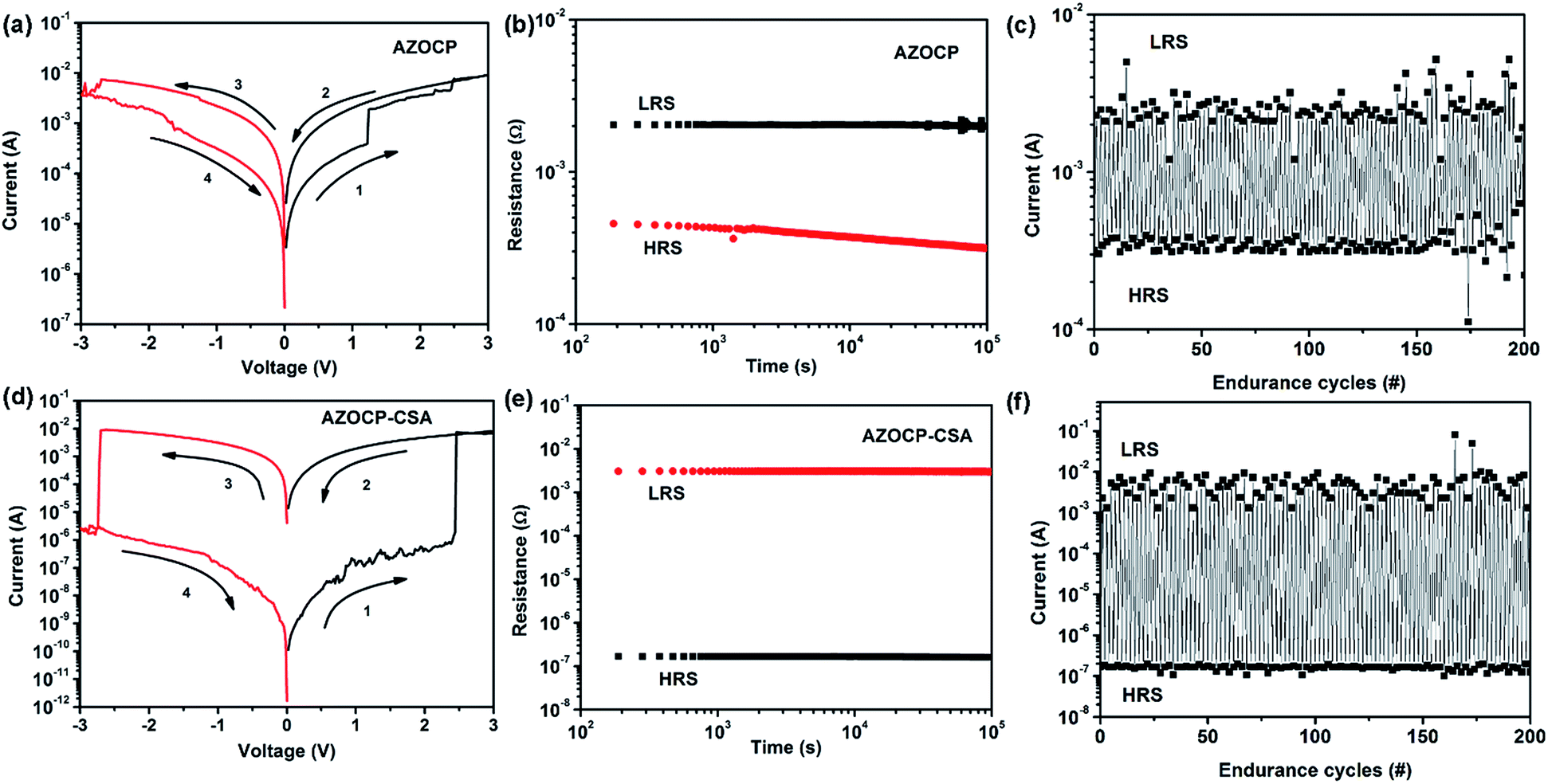 | ||
| Fig. 5 Current–voltage (I–V) characteristics of Au/AZOCP/ITO and Au/AZOCP–CSA/ITO memory device (a and d); the effect of retention time (b and e) and the endurance cycles of Au/AZOCP/ITO and Au/AZOCP–CSA/ITO memory device under a constant stress of −1.0 V (c and f). | ||
Compared with the Au/AZOCP/ITO devices, the Au/AZOCP–CSA/ITO devices exhibited much smoother I–V characteristics during the resistive-switching process. As shown in Fig. 5d, the Au/AZOCP–CSA/ITO memory cells was in the HRS. When a positively biased potential sweep from 0 to 3 V was applied (sweep 1 in the Fig. 5d), the current increased consecutively from ∼10−6 A to ∼10−2 A, indicating that the device had been set to the LRS by the positive forward sweep. This “Write” (or “SET”) process was confirmed by the subsequent positive backward potential sweep from 3 to 0 V (sweep 2). In the following sweep from 0 to −3 V (sweep 3), the device abruptly decreased to the HRS. The Au/AZOCP–CSA/ITO devices can remains at the HRS by applying a negatively-biased voltage sweeping from −3 V to 0 V (sweeps 4). The value of the ON/OFF ratio for AZOCP–CSA-base devices was 105. Compared with the Au/AZOCP/ITO device, the AZOCP–CSA-base device has higher ON/OFF ratio, which is helpful to avoid false programming and error readout problems.
The retention times of the RLRS and the RHRS of the Au/AZOCP/ITO and Au/AZOCP–CSA/ITO device are shown in Fig. 5b and e. Under a constant stress of −1 V, AZOCP–CSA-based devices is observed more stability and no significant degradation in current for any of the states at least 105 s during the readout test. The endurance cycles of the Au/AZOCP–CSA/ITO device is much better than that of the Au/AZOCP/ITO device (as shown in Fig. 5c and f). The rewritable cycles (write–read–erase–read) of AZOCP–CSA-base device could be repeated over 200 times and almost no attenuation.
Memory mechanism
First of all, we choose Au electrode instead of Al electrode in order to avoid the formation of Al2Ox layer to affect the bistable memory performance.43 Moreover, the low LRS current density (10−2 A) is lower than current compliance (10−1 A) and the lack of necessary forming process or device breakdown in our devices are speculated to exclude the metal filament mechanism.44 To explain the current flow by adding an applied voltage, carrier transport mechanisms were analyzed through isothermal I–V correction. The related physical models have been described by follows: space-charge-limited conduction (SCLC; including ohmic conduction (I ≈ V) and child's law region (I ≈ V2)). As shown in Fig. 6, the I–V curve of LRS and HRS of Au/AZOCP/ITO devices satisfactorily fits the classical SCLC model. However, there are two different regions in the HRS of Au/AZOCP–CSA/ITO device. At the low voltage region of HRS, standard ohmic conduction model is dominated. It suggests a linear relationship of I–V curve (I ∝ V1.15), whereas in the high voltage region, the current density shows the square dependence of the voltage that confirms the child's mode (I ∝ V2.37) (Fig. 6b). The SCLC can determine the charge carrier injection and hopping through the thin films in HRS. When the voltage continue to increase and the current density increases exponentially to LRS. The charge injection/conduction in LRS (inset of Fig. 6b) is well fitted to the SCLC model.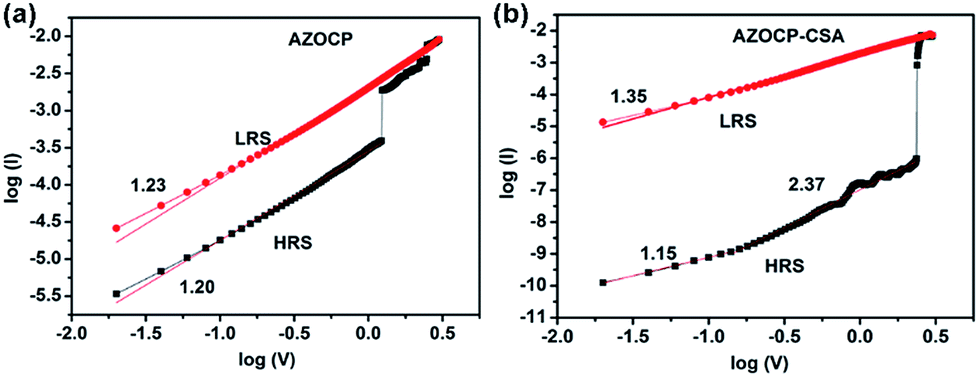 | ||
| Fig. 6 Experimental and fitted I–V curves of Au/AZOCP/ITO (a) and Au/AZOCP–CSA/ITO (b) memory device in both the ON and OFF states during the SET process. | ||
To better understand the electronic process occurring inside the thin film. Theoretical calculations were performed using density-functional program DMol3.45,46 The hybrid functional B3LYP,47,48 together with a double numerical plus polarization (DNP) basis set, was employed. For simplification, the camphorsulfonic acid in AZOCP–CSA are omitted in the computational models; this representation has little impact on the calculation results. The calculated results on HOMO, LUMO and energy gap (Egap) are listed in Table S1.† Compared the Egap of AZOCP and AZOCP–CSA, the AZOCP–CSA has lower Egap, which make charge carrier migration easier.
The molecular orbital (HOMO−2, HOMO−1, HOMO, LUMO, LUMO+1) and the electrostatic surface potential (ESP) of AZOCP and AZOCP–CSA molecules were plotted, and shown in Fig. 7. The ESP plots of AZOCP and AZOCP–CSA molecules both show an open channel along the molecular backbone with continuous positive electrostatic potential, providing a path to allow charge carrier migration. For AZOCP molecule, electrons transit readily from HOMO orbital to LUMO orbital under external electric field, forming the locally excited state. Meanwhile, electrons in HOMO−2 can overcome the energy barrier between HOMO−2 and HOMO and fill the generated holes in HOMO, followed by the spontaneous electron transition from HOMO−1 to HOMO−2. As a result, a charge-transfer (CT) interaction can occur in AZOCP molecule between the electron donor moieties and the electron acceptor moieties. For AZOCP–CSA molecule, a similar charge transfer process is also formed. However, the salification enhances the electron-withdrawing ability of acceptor moiety, so the HOMO and LUMO orbitals of AZOCP–CSA molecules show more obvious separation in ground state, with HOMO mostly localizing on donor areas and LUMO mostly localizing on acceptor areas.
 | ||
| Fig. 7 The HOMO−2, HOMO−1, HOMO, LUMO, LUMO+1 orbital and molecular ESP from DFT simulation result of AZOCP and AZOCP–CSA. | ||
We speculated that the mechanism of the resistance-switching effects of the AZOCP and AZOCP–CSA devices could be the electric-field-induced charge transfer effect. Without external electric field, the electrons in AZOCP and AZOCP–CSA molecules were stable, and the as-fabricated device was in the HRS. When applied a positive bias, the charge transport pathways will form, and will switch the Au/AZOCP or AZOCP–CSA/ITO device from the HRS to the LRS. However, the salification can significantly enhance the electron-withdrawing ability of pyridine group,49,50 resulting in a stronger intramolecular CT in AZOCP–CSA. There have been reported that changing the charge transfer ability of D–A molecules can tune memory effects.51 So the better performance (larger ON/OFF ratio and more stable of each electric conductive state) of the AZOCP–CSA-based device could due to the stronger intramolecular CT effect. Application of a reverse positive bias to the AZOCP-based or AZOCP–CSA-based device can extract electrons from the acceptor moieties and program the device back to the HRS.52 The nonvolatile nature of the LRS is due to the intensive electron delocalization in the acceptor moieties stabilized the conductive CT state.53
In order to further investigate the effect of salification on the device performance. We synthesized another new molecule with pyridine group: 3-(2-(dodecyloxy)-4-((4-nitrophenyl)diazenyl) phenyl)-1-(pyridin-4-yl)prop-2-en-1-one (AZOCP2) and its camphorsulfonic acid salification compound (AZOCP2–CSA) through the similar method mentioned above (as shown in Scheme S1†). Further we studied the resistive memory properties of this compound based memories (as shown in Fig. S6†). After salification the compound AZOCP2–CSA was also shown a higher ON/OFF ratio (from ∼103 to ∼104) and more stablable retention times (Fig. S6†). So we think that this kind of organic molecule with pyridine group system might improve the resistive memory properties by salification.
Conclusions
In summary, we designed and synthesised a new small molecule AZOCP and its salification compound AZOCP–CSA. From the UV-vis absorbance spectra, AFM and XRD measurement, the AZOCP–CSA molecule shows a stronger π–π conjugation and leads to a more ordered stacking nanocrystalline film. Moreover, the Au/AZOCP–CSA/ITO memory device has a higher ON/OFF ratio and more stablable retention times and longer rewritable cycles than the Au/AZOCP/ITO memory device. The salification can promote the transport of charge carriers to promote memory performance in resistive switching. We demonstrate a very simple but effective strategy to improve organic resistive memory devices with good performance.Acknowledgements
The authors thank the Chinese Natural Science Foundation (21336005, 21176164 and 21371128), Qing-Lan Project of Jiangsu Province and the Major Project of Department of Education in Jiangsu Province (15KJA150008).References
- Y. Chen, G. Liu, C. Wang, W. Zhang, R.-W. Li and L. Wang, Mater. Horiz., 2014, 1, 489–506 RSC.
- X. Marti, I. Fina, C. Frontera, J. Liu, P. Wadley, Q. He, R. J. Paull, J. D. Clarkson, J. Kudrnovsky, I. Turek, J. Kunes, D. Yi, J. H. Chu, C. T. Nelson, L. You, E. Arenholz, S. Salahuddin, J. Fontcuberta, T. Jungwirth and R. Ramesh, Nat. Mater., 2014, 13, 367–374 CrossRef CAS PubMed.
- C. D. Wright, Y. Liu, K. I. Kohary, M. M. Aziz and R. J. Hicken, Adv. Mater., 2011, 23, 3408–3413 CrossRef CAS PubMed.
- J. J. Yang, F. Miao, M. D. Pickett, D. A. A. Ohlberg, D. R. Stewart, C. N. Lau and R. S. Williams, Nanotechnology, 2009, 20, 215201–215209 CrossRef PubMed.
- H. J. Koo, J. H. So, M. D. Dickey and O. D. Velev, Adv. Mater., 2011, 23, 3559–3564 CrossRef CAS PubMed.
- E. N. Oskoee and M. Sahimi, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 031105–031108 CrossRef PubMed.
- Q. D. Ling, D. J. Liaw, C. Zhu, D. S. H. Chan, E. T. Kang and K. G. Neoh, Prog. Polym. Sci., 2008, 33, 917 CrossRef CAS.
- S. Song, B. Cho, T. W. Kim, Y. Ji, M. Jo, G. Wang, M. Choe, Y. H. Kahng, H. Hwang and T. Lee, Adv. Mater., 2010, 22, 5048–5052 CrossRef CAS PubMed.
- B. B. Cui, J. H. Tang, J. Yao and Y. W. Zhong, Angew. Chem., Int. Ed., 2015, 54, 9192–9197 CrossRef CAS PubMed.
- C. T. Poon, D. Wu, W. H. Lam and V. W. Yam, Angew. Chem., Int. Ed., 2015, 54, 10569–10573 CrossRef CAS PubMed.
- S. E. Savel'ev, A. S. Alexandrov, A. M. Bratkovsky and R. S. Williams, Nanotechnology, 2011, 22, 254011–254017 CrossRef PubMed.
- X. Sun, G. Li, L. Ding, N. Yang and W. Zhang, Appl. Phys. Lett., 2011, 99, 072101–072103 CrossRef.
- J. Fang, H. You, J. Chen, J. Lin and D. Ma, Inorg. Chem., 2006, 45, 3701–3704 CrossRef CAS PubMed.
- J. Xiao, Z. Yin, H. Li, Q. Zhang, F. Boey, H. Zhang and Q. Zhang, J. Am. Chem. Soc., 2010, 132, 6926–6928 CrossRef CAS PubMed.
- C. Simao, M. Mas-Torrent, J. Casado-Montenegro, F. Oton, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2011, 133, 13256–13259 CrossRef CAS PubMed.
- B. Hu, C. Wang, J. Wang, J. Gao, K. Wang, J. Wu, G. Zhang, W. Cheng, B. Venkateswarlu, M. Wang, P. S. Leea and Q. Zhang, Chem. Sci., 2014, 5, 3404 RSC.
- S. Miao, H. Li, Q. Xu, Y. Li, S. Ji, N. Li, L. Wang, J. Zheng and J. Lu, Adv. Mater., 2012, 24, 6210–6215 CrossRef CAS PubMed.
- P. Gu, J. He, G. Long, C. Wang, W. Chen, Q. Xu, J. Lu and Q. Zhang, J. Mater. Chem. C, 2015, 3, 3167–3172 RSC.
- Y. Zhang, H. Zhuang, Y. Yang, X. Xu, Q. Bao, N. Li, H. Li, Q. Xu, J. Lu and L. Wang, J. Phys. Chem. C, 2012, 116, 22832–22839 CAS.
- Z. Liu, E. Shi, Y. Wan, N. Li, D. Chen, Q. Xu, H. Li, J. Lu, K. Zhang and L. Wang, J. Mater. Chem. C, 2015, 3, 2033–2039 RSC.
- Z. Liu, J. He, H. Zhuang, H. Li, N. Li, D. Chen, Q. Xu, J. Lu, K. Zhang and L. Wang, J. Mater. Chem. C, 2015, 3, 9145–9153 RSC.
- J. S. Lee, Y. M. Kim, J. H. Kwon, J. S. Sim, H. Shin, B. H. Sohn and Q. Jia, Adv. Mater., 2011, 23, 2064–2068 CrossRef CAS PubMed.
- S. J. Liu, P. Wang, Q. Zhao, H. Y. Yang, J. Wong, H. B. Sun, X. C. Dong, W. P. Lin and W. Huang, Adv. Mater., 2012, 24, 2901–2905 CrossRef CAS PubMed.
- C. Ye, Q. Peng, M. Li, J. Luo, Z. Tang, J. Pei, J. Chen, Z. Shuai, L. Jiang and Y. Song, J. Am. Chem. Soc., 2012, 134, 20053–20059 CrossRef CAS PubMed.
- C. Wang, B. Hu, J. Wang, J. Gao, G. Li, W.-W. Xiong, B. Zou, M. Suzuki, N. Aratani, H. Yamada, F. Huo, P. S. Lee and Q. Zhang, Chem.–Asian J., 2015, 10, 116–119 CrossRef PubMed.
- G. Li, K. Zheng, C. Wang, K. S. Leck, F. Hu, X. W. Sun and Q. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 6458 CAS.
- P. Y. Gu, F. Zhou, J. Gao, G. Li, C. Wang, Q. F. Xu, Q. Zhang and J. M. Lu, J. Am. Chem. Soc., 2013, 135, 14086 CrossRef CAS PubMed.
- L. Xie, Q. Ling, X. Hou and W. Huang, J. Am. Chem. Soc., 2008, 130, 2120–2121 CrossRef CAS PubMed.
- S. K. Hwang, J. M. Lee, S. Kim, J. S. Park, H. I. Park, C. W. Ahn, K. J. Lee, T. Lee and S. O. Kim, Nano Lett., 2012, 12, 2217–2221 CrossRef CAS PubMed.
- J. H. A. Smits, S. C. J. Meskers and R. J. Janssen, Adv. Mater., 2005, 17, 1169–1173 CrossRef CAS.
- F. Verbakel, S. C. J. Meskers and R. J. Janssen, Chem. Mater., 2006, 18, 2707–2712 CrossRef CAS.
- S. Miao, Y. Zhu, Q. Bao, H. Li, N. Li, S. Ji, Q. Xu, J. Lu and L. Wang, J. Phys. Chem. C, 2014, 118, 2154–2160 CAS.
- A. Tilborg, B. Norberg and J. Wouters, Eur. J. Med. Chem., 2014, 74, 411–426 CrossRef CAS PubMed.
- M. Wu, Z. W. Wang, Y. X. Liu, H. B. Song, A. Zhang, L. H. Meng and Q. M. Wang, New J. Chem., 2013, 37, 1817–1822 RSC.
- B. Hu, X. Zhu, X. Chen, L. Pan, S. Peng, Y. Wu, J. Shang, G. Liu, Q. Yan and R. W. Li, J. Am. Chem. Soc., 2012, 134, 17408–17411 CrossRef CAS PubMed.
- J. Liu, J. W. Y. Lam and B. Z. Tang, J. Inorg. Organomet. Polym., 2009, 19, 249–285 CrossRef CAS.
- F. L. Ye, P. Y. Gu, F. Zhou, H. F. Liu, X. P. Xu, H. Li, Q. F. Xu and J. M. Lu, Polymer, 2013, 54, 3324–3333 CrossRef CAS.
- H. Li and A. G. McDonald, Ind. Crops Prod., 2014, 62, 67–76 CrossRef CAS.
- H. Li, G. Sivasankarapillai and A. G. McDonald, Ind. Crops Prod., 2015, 67, 143–154 CrossRef CAS.
- S. Kim, T. K. An, J. Chen, I. Kang, S. H. Kang, D. S. Chung, C. E. Park, Y. H. Kim and S. K. Kwon, Adv. Funct. Mater., 2011, 21, 1616–1623 CrossRef CAS.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed.
- L. Li, P. Gao, K. C. Schuermann, S. Ostendorp, W. Wang, C. Du, Y. Lei, H. Fuchs, L. Cola and L. Chi, J. Am. Chem. Soc., 2010, 132, 8807–8809 CrossRef CAS PubMed.
- A. D. Yu, T. Kurosawa, Y. H. Chou, K. Aoyag, T. Higashihara, M. Ueda, C. L. Liu and W. C. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 4921–4929 CAS.
- F. Zhuge, B. Hu, C. He, X. Zhou, Z. Liu and R. W. Li, Carbon, 2011, 49, 3796–3802 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. Stephens, F. Devlin, C. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. Moller, C. Perlov, W. Jackson, C. Taussig and S. R. Forrest, Nature, 2003, 426, 166 CrossRef PubMed.
- R. Sim, M. Y. Chan, A. S. W. Wong and P. S. Lee, Org. Electron., 2011, 12, 185 CrossRef CAS.
- C. L. Liu and W. C. Chen, Polym. Chem., 2011, 2, 2169 RSC.
- B. Zhang, G. Liu, Y. Chen, L. J. Zeng, C. X. Zhu, K. G. Neoh, C. Wang and E. T. Kang, Chem.–Eur. J., 2011, 17, 13646–13652 CrossRef CAS PubMed.
- W. Zhang, C. Wang, G. Liu, X. Zhu, X. Chen, L. Pan, H. Tan, W. Xue, Z. Ji, J. Wang, Y. Chen and R. W. Li, Chem. Commun., 2014, 50, 11856–11858 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data, Fig. S1–S4. Thermogravimetric analysis of the both molecules, Fig. S5. Current–voltage (I–V) characteristics, the endurance cycles and the effect of retention time of AZOCP2 and AZOCP–CSA, Fig. S6. See DOI: 10.1039/c5ra25099d |
| This journal is © The Royal Society of Chemistry 2016 |