
Fabrication and testing of zirconium-based nanoparticle-doped activated carbon fiber for enhanced arsenic removal in water†
Abstract
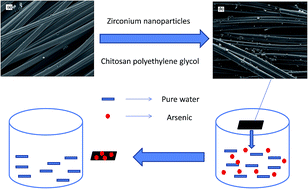
Arsenic contamination in groundwater has raised great concern in many countries around the world. In this study, a new activated carbon fiber (ACF) based material targeting the simultaneous removal of arsenic and natural organic matter was developed. The ACF, as a support material, was modified by doping with zirconium-based nanoparticles (NP) with chitosan as a linking polymer. The fabrication of Zr-based NP doped ACF was optimized by a L9 (3)4 orthogonal experimental design approach. The adsorption kinetics study showed that the adsorption equilibrium was established within 30 h. The adsorption increased as the solution pH was decreased; the optimal pH for adsorption was 3.0. The experimental data were better described by the Langmuir equation than the Freundlich equation; the maximum adsorption capacity of 21.7 mg As per g was achieved at pH 3.0. The adsorption was not reduced in the presence of carbonate. The presence of fluoride and phosphate had some negative effects on the adsorption. The arsenic uptake was however greatly retarded by the silicate. The fixed-bed column filtration experiment demonstrated that the sorbent had 570.4 bed volumes, to meet the maximum contaminant level of 10 µg L−1 when treating simulated arsenic contaminated water with an initial concentration of 106 µg L−1. The XPS analysis indicated that the adsorption of arsenate was mainly associated with an ion-exchange reaction between hydrogen sulfate and arsenate ions.
 Please wait while we load your content...
Please wait while we load your content...

