
Organocatalytic construction of spirooxindole naphthoquinones through Michael/hemiketalization using L-proline derived bifunctional thiourea†
V. Pratap Reddy Gajulapalli,
Kanduru Lokesh,
Manjunatha Vishwanath and
Venkitasamy Kesavan*
Chemical Biology Laboratory, Department of Biotechnology, Bhupat and Jyothi Mehtha School of Biosciences, Indian Institute of Technology Madras, Chennai-600036, India. E-mail: vkesavan@iitm.ac.in; Fax: +91-44-2257-4102; Tel: +91-44-2257-4124
First published on 22nd January 2016
Abstract
Michael/hemiketalization of 2-hydroxy-1,4-naphthoquinone to oxindole ketoester was studied using a series of chiral bifunctional organocatalysts. Good yields (up to 91%) and excellent enantioselectivities (up to 98%) were achieved by using a proline derived thiourea catalyst. This method provides an elegant synthetic route to access oxindole containing naphthoquinone derivatives.
In chemical biology, researchers employ chemical probes to explore biological systems. Due to the unmet need of identifying small molecule binding partners for biological targets, access to compound collections with wide biological activities is highly desirable. This need for novel molecules to decipher cellular processes inspires synthetic chemists to construct new molecules around privileged structural motifs.1 Creation of chiral molecular complexity around biologically active scaffolds is an ongoing challenge for synthetic organic chemists.2 Enantiomerically pure spirooxindoles are being used to unravel biological pathways in recent years.3a,b,d,f Hence, heterocyclic spirooxindoles are widely targeted by synthetic chemists due to their prevalent presence in various pharmacologically active scaffolds and natural products.3c,e,g,h Intrigued by the biological activities of spirooxindoles, various groups have developed scaffold inspired enantioselective synthesis of novel spirooxindoles.4 Development of these efficient methodologies make significant contribution in guiding the discovery of new chemical entities.
A large number of natural products and biologically active molecules contain quinine and naphthoquinone structural motifs.5 Hence these derivatives have been explored for their antitumor,6,7 antifungal,8 molluscicidal,9 anti-inflammatory,10 trypanocidal,11 and leishmanicidal biological activities.12 Intrigued by the pharmacological activities of spirooxindole motifs and naphthoquinone derivatives, we wish to synthesize molecules which comprise both of these pharmacophores. The hybrid molecule, spirooxindole naphthoquinone 3, may inherit biological activities of both spirooxindole and naphthoquinone skeletons. Synthesis of 3 could be easily achieved by tandem Michael/hemiketalization of ketoester 1 with 2-hydroxy-1,4- naphthoquinone 2. However identification of suitable catalyst to facilitate this transformation is a formidable task.
Organocatalysts are emerging as a method of choice to perform C–C bond formation for the efficient enantioselective assembly of complex molecular architecture enantioselectively.13 Especially chiral bifunctional thioureas play a vital role in various asymmetric transformations in C–C bond formation reactions.14 In our continuing efforts in identifying new catalysts for enantioselective transformations, we have developed a novel bifunctional thiourea and uncovered its potential in various asymmetric transformations.15 Having demonstrated the capability of chiral proline thiourea in catalyzing C–C bond formation, we sought to explore the same catalyst for the assembly of novel spirooxindole naphthaquinones 3.
On the basis of our hypothesis (Scheme 1), ketoester 1 was reacted with 2-hydroxy-1,4- naphthoquinone 2 in the presence of various bases and bifunctional organocatalysts (5 mol%) at room temperature in dichloromethane. The results are summarized in the Table 1.
 | ||
| Scheme 1 Strategy for enanioselective synthesis of spirooxindole naphthoquinones. | ||
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Yieldb (%) | eec (%) |
| a The reactions were carried out with 1a (0.1 mmol), 2 (0.1 mmol), and catalyst 4 (0.005 mmol) in 1 ml of DCM at 25 °C.b Isolated yield.c Determined by chiral HPLC stationary phase. A minus sign indicate opposite enantiomer. | ||||
| 1 | 4a | 2 | 81 | −40 |
| 2 | 4b | 2 | 84 | −70 |
| 3 | 4c | 2 | 82 | −60 |
| 4 | 4d | 2 | 82 | 27 |
| 5 | 4e | 2 | 81 | 37 |
| 6 | 4f | 2 | 86 | −27 |
| 7 | 4g | 5 | 81 | 98 |
| 8 | 4h | 10 | — | — |
| 9 | 4i | 5 | 86 | −72 |
| 10 | 4j | 4 | 80 | −84 |
| 11 | 4k | 3 | 81 | −90 |
| 12 | 4l | 3 | 83 | 83 |
| 13 | 4m | 5 | 79 | −75 |
| 14 | 4n | 5 | 84 | −93 |
It is evident from these observations that although chiral bases efficiently catalyzed the reaction to afford the expected spirooxindole naphthoquinone 3a in very good yields, the enantioselectivity of the reaction was found to be poor. Only quinidine 4b yielded the expected product with a moderate enantiomeric excess of 70% (Table 1, entry 2). In screening bifunctional organocatalysts, we decided to firstly evaluate L-proline derived thiourea 4g developed by our group. To our delight, organocatalyst 4g efficiently transformed the reactants into spirooxindole naphthoquinone 3a in very good yield and excellent enantioselectivity (Table 1, entry 7). To demonstrate the requirement of stereogenic centre at the carbon bearing thiourea moiety, catalysts 4h and 4i were evaluated in the synthesis of spirooxindole naphthoquinone 3a.
Diphenyl substitutions on stereogenic center suppressed the catalytic efficiency of 4h entirely and no product formation was observed (Table 1, entry 8). Although employment of organocatalyst 4i furnished the expected product in 86% yield and the enantimeric excess was only moderate (Table 1, entry 9). HPLC profile proved that the opposite stereoisomer was obtained when organocatalyst 4i was employed. This shows that stereochemical outcome in assembling spirooxindole naphthaquinones, is controlled by the newly created stereogenic carbon which bears thiourea moiety.
The efficiency of widely used thioureas was also examined in constructing spirooxindole naphthoquinone 3a (Table 1, entries 10–12). Takemoto 4j, quinine 4k and quinidine 4l derived thioureas catalysed the transformation to afford the expected product 3a in good yields. The enantioselectivity of the product 3a was found to be little lower in case of Takemoto thiourea 4j and quinidine thiourea 4l (Table 1, entries 10 and 11), when compared with quinine derived thiourea 4k (Table 1, entry 12). Formation of spirooxindole naphthoquinone 3a was carried out in the presence of thioureas containing pyrrolidine ring 4m and 4n (Table 1, entries 13–14). The enantioselectivity got reduced, when thiourea 4m was employed as the catalyst (Table 1, entry 13). Valinol derived thiourea 4n furnished the product 3a with 84% yield and 93% ee (Table 1, entry 14). It is evident from Table 1 that proline derived bifunctional organocatalyst 4g is the most effective catalyst in furnishing the expected product with very good yield and excellent enantioselectivity (98% ee).
Encouraged by the successful construction of optically active spirooxindole naphthoquinone 3a, we probed the scope of this reaction with the range of oxindole ketoesters 1. We are glad to observe that irrespective of N1 substitution, the expected products were obtained with no significant loss of yield and enantioselectivity. N-benzyl 1b, N-propargyl 1c and N-allyl 1d substitutions are well tolerated in the assembly of optically active spirooxindole naphthaquinones (Table 2 entries 2–4). This suggests that organocatalyst 4g may interact with substrate 1 in the vicinity of ketoester part preferably than oxindole moiety (Scheme 1).
|
||||
|---|---|---|---|---|
| Entry | R1, R2 | Product | Yieldb (%) | eec (%) |
| a The reactions were carried out with 1 (0.1 mmol), 2 (0.1 mmol), and catalyst 4g (0.005 mmol) in 1 ml of DCM at rt for 5 h.b Isolated yield.c Determined using chiral HPLC stationary phase.d Reaction was carried out at 5 °C. | ||||
| 1 | Me, H 1a | 3a | 84 | 98 |
| 2 | Bn, H 1b | 3b | 85 | 97 |
| 3 | Propargyl, H 1c | 3c | 81 | 93 |
| 4 | Allyl, H 1d | 3d | 86 | 97 |
| 5 | Me, 5-F 1e | 3e | 91 | 96 |
| 6 | Me, 5-Cl 1f | 3f | 88 | 98 |
| 7 | Me, 5-Br 1g | 3g | 91 | 96 |
| 8 | Me, 5-I 1h | 3h | 90 | 98 |
| 9 | Me, 5-OMe 1i | 3i | 82 | 95 |
| 10 | Me, 5-OCF3 1j | 3j | 86 | 90 |
| 11 | Me, 5,7-dimethyl 1k | 3k | 81 | 89 |
| 12 | Me, 7F 1l | 3l | 81 | 94 |
| 13 | H, H 1m | 3m | 78 | 50(60)d |
Under the optimized reaction conditions the generality of our protocol was tested using various 5-substituted oxindole ketoesters having electron-withdrawing, electron-donating and other substituents. We are pleased to note that the substitution pattern on aromatic ring had limited influence on enantioselectivity and there was no negative effect on yield. Various 5-halo substituted oxindole ketoesters 1e–1h afforded the respective products 3e–3h in excellent yields and enantioselectivities (Table 2, entries 5–8). Irrespective of electronic nature of 5-substituents on the aryl ring of oxindole ketoesters 1i and 1j, the completion of reactions were confirmed within 12 hours in very good yields and enantioselectivity (Table 2, entries 9 and 10).
Presence of 5,7-dimethyl 1k and 7 F 1l of oxindole ketoesters did not affect enantioselectivity and the desired products 3k and 3l were isolated in good yields (Table 2, entries 11 and 12). Oxindole ketoester which is devoid of substitutions of N1 atom underwent Michael addition followed by intramolecular cyclisation to yield the product 3m in 78% yield and 50% ee. Lowering of temperature to 5 °C, the enantioselectivity was improved to 60%. Further lowering the temperature did not result in expected improvement of enantioselectivity. This result suggests that when there is no substituent on N1 atom, there may be a competitive binding of organocatalyst with oxindole moiety of ketoester as well as interacting with ketoester portion. Activation of both reactants may not be possible if the catalyst interacts with oxindole moiety instead of ketoester portion. Thus, the moderate ee observed for oxindole ketoester 1m may be attributed to the competitive binding (Table 2, entry 13).
In order to show the potentiality of this methodology, the reaction was carried out with 2 mmol of 1a, in the presence of 5 mol% of catalyst 4g under the optimized conditions. The desired product 3a was afforded with no loss in yield and 88% enantioselectivity. Thus we established an asymmetric protocol to construct a wide range of spiro oxindole frameworks with excellent enantioselectivity.
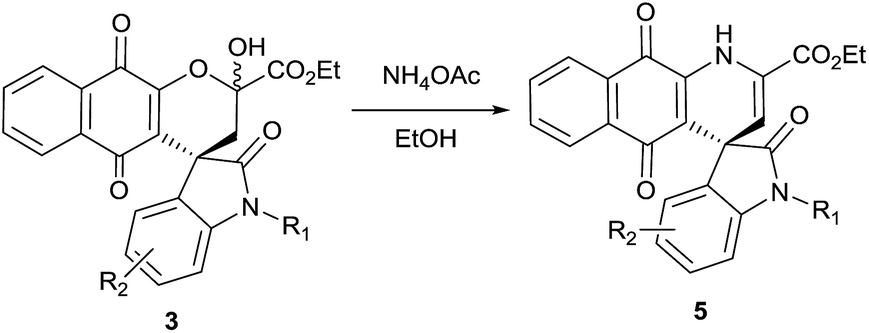
Our efforts to crystallize synthesized compounds 3 did not yield fruitful result. Hence to demonstrate the potential application of these derivatives 3 and to determine absolute configuration at spiro centre, the compounds 3 were treated with ammonium acetate at ambient temperature in ethanol to yield spirooxindole dihydropyridine naphthoquinones derivatives 5 (Table 3, entries 1–5). To our delight structure of product 5b was confirmed by single-crystal X-ray analysis and absolute configuration was determined to be “R” (Fig. 1).
|
||||
|---|---|---|---|---|
| Entry | R1, R2 | Product | Yieldb (%) | eec (%) |
| a The reactions were carried out with 3 (0.1 mmol), NH4OAc (0.15 mmol) in ethanol at rt for 6 h.b Isolated yield.c Determined using chiral HPLC stationary phase. | ||||
| 1 | Allyl, H, 3d | 5a | 89 | 89 |
| 2 | Me, 5-I, 3h | 5b | 80 | 96 |
| 3 | Me, 5,7-dimethyl, 3k | 5c | 85 | 80 |
| 4 | Me, 7-F, 3l | 5d | 90 | 98 |
| 5 | Me, 5-Cl, 3f | 5e | 82 | 97 |
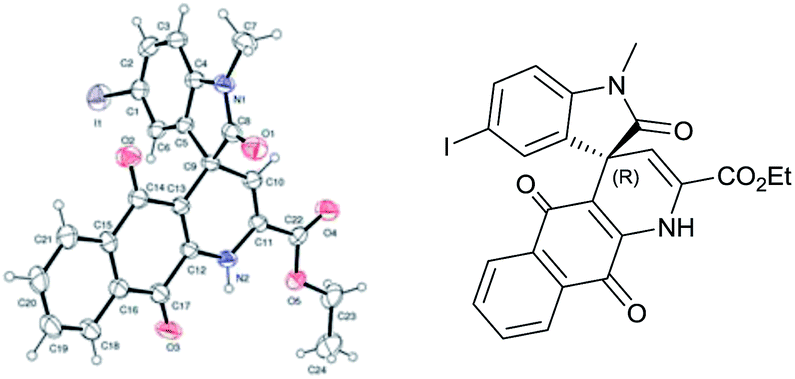 | ||
| Fig. 1 X-ray crystal structure of compound 5b. | ||
The reaction mechanism is proposed via Michael addition followed by hemiketalization catalyzed by bifunctional catalyst 4g (Fig. 2). Oxindole ketoester 1 is activated to nucleophilic attack through double hydrogen-bonding interactions with the thiourea group of catalyst 4g and tertiary amine of catalyst deprotonates 2-hydroxynaphthaquinone 2. The resulting ammonium ion forms another hydrogen bond with the enolate of 2. The electron-rich α-carbon atom of 2-hydroxynaphthoquinone 2 predominantly attacks the Re-face of the electron-deficient oxindole ketoester 1 to generate the Micheal adduct intermediate A and subsequent intramolecular cyclization afforded the spirooxindole naphthoquinones 3 with “R” as absolute configuration at spirocentre of oxindole moiety (Fig. 2).
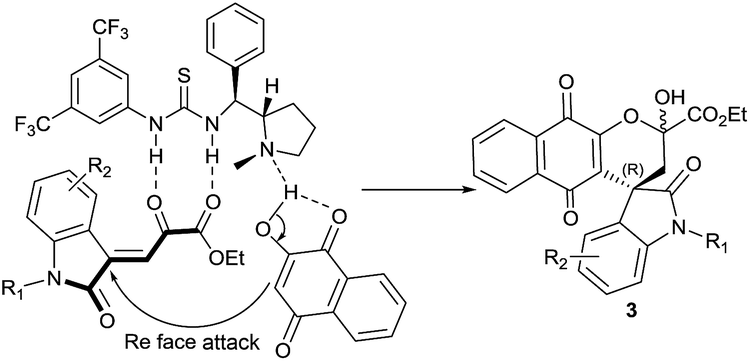 | ||
| Fig. 2 Plausible mechanism for Michael/hemiketalization reaction. | ||
Conclusions
In conclusion, we have demonstrated highly efficient Michael/hemiketalization reaction for the construction of spirooxindole naphthoquinones with good yields (78–91%) and excellent enantioselectivities (up to 98% ee). To the best our knowledge, no enantioselective spirooxindole naphthoquinones have been reported previously. Proline based thiourea 4g was identified as the efficient catalyst for this transformation.Acknowledgements
We acknowledge CEFIPRA, New Delhi (Grant No. 4803-4) for financial assistance, GVP thanks Council of Scientific and Industrial Research for research fellowship. We thank Dr C. Baby at Sophisticated Analytical Instrument Facility (SAIF), IIT Madras for NMR analysis. We thank Dr Sudha devi for crystallographic analysis SAIF-IIT Madras for spectral analysis.Notes and references
- (a) R. Kneller, Nat. Rev. Drug Discovery, 2010, 9, 955 CrossRef CAS; (b) J. Kim, H. Kim and S. B. Park, J. Am. Chem. Soc., 2014, 136, 14629–14638 CrossRef CAS PubMed.
- (a) W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 13 Search PubMed; (b) J. J. Badillo, N. V. Hanhan and A. K. Franz, Curr. Opin. Drug Discovery Dev., 2010, 13, 758–776 CAS.
- (a) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed; (b) M. Rottmann, C. McNamara, B. K. S. Yeung, M. C. S. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. E. Gonzalez-Paez, S. B. Lakshminarayana, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H.-P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175–1180 CrossRef CAS PubMed; (c) A. Lauria, M. Tutone, M. Ippolito, L. Pantano and A. M. Almerico, Curr. Med. Chem., 2010, 17, 3142–3154 CrossRef CAS PubMed; (d) Y. Zhao, L. Liu, W. Sun, J. Lu, D. McEachern, X. Li, S. Yu, D. Bernard, P. Ochsenbein, V. Ferey, J.-C. Carry, J. R. Deschamps, D. Sun and S. Wang, J. Am. Chem. Soc., 2013, 135, 7223–7234 CrossRef CAS PubMed; (e) Y. Zhao, L. Liu, W. Sun, J. Lu, D. McEachern, X. Li, S. Yu, D. Bernard, P. Ochsenbein, V. Ferey, J.-C. Carry, J. R. Deschamps, D. Sun and S. Wang, J. Am. Chem. Soc., 2013, 135, 7223–7234 CrossRef CAS PubMed; (f) Y. Kia, H. Osman, R. S. Kumar, V. Murugaiyah, A. Basiri, S. Perumal and I. A. Razak, Bioorg. Med. Chem. Lett., 2013, 23, 2979–2983 CrossRef CAS PubMed; (g) R. Narayan, M. Potowski, Z.-J. Jia, A. P. Antonchick and H. Waldmann, Acc. Chem. Res., 2014, 47, 1296–1310 CrossRef CAS PubMed; (h) Y. Zhao, A. Aguilar, D. Bernard and S. Wang, J. Med. Chem., 2015, 58, 1038–1052 CrossRef CAS PubMed.
- (a) K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qiu, Y. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller, Y. Tomita, D. A. Parrish, J. R. Deschamps and S. Wang, J. Am. Chem. Soc., 2005, 127, 10130–10131 CrossRef CAS PubMed; (b) X.-H. Chen, Q. Wei, S.-W. Luo, H. Xiao and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 13819–13825 CrossRef CAS PubMed; (c) X. Jiang, Y. Cao, Y. Wang, L. Liu, F. Shen and R. Wang, J. Am. Chem. Soc., 2010, 132, 15328–15333 CrossRef CAS PubMed; (d) A. P. Antonchick, C. Gerding-Reimers, M. Catarinella, M. Schuermann, H. Preut, S. Ziegler, D. Rauh and H. Waldmann, Nat. Chem., 2010, 2, 735–740 CrossRef CAS PubMed; (e) X. Zhou, T. Xiao, Y. Iwama and Y. Qin, Angew. Chem., Int. Ed., 2012, 51, 4909–4912 CrossRef CAS PubMed; (f) X. Jiang, Y. Sun, J. Yao, Y. Cao, M. Kai, N. He, X. Zhang, Y. Wang and R. Wang, Adv. Synth. Catal., 2012, 354, 917–925 CrossRef CAS; (g) D. A. Mundal and R. Sarpong, Org. Lett., 2013, 15, 4952–4955 CrossRef CAS PubMed; (h) X.-M. Shi, W.-P. Dong, L.-P. Zhu, X.-X. Jiang and R. Wang, Adv. Synth. Catal., 2013, 355, 3119–3123 CrossRef CAS; (i) M. M. M. Santos, Tetrahedron, 2014, 70, 9735–9757 CrossRef CAS; (j) Y.-K. Liu, M. Nappi, E. Arceo, S. Vera and P. Melchiorre, J. Am. Chem. Soc., 2011, 133, 15212–15218 CrossRef CAS PubMed; (k) B. Tan, G. Hernandez-Torres and C. F. Barbas, J. Am. Chem. Soc., 2011, 133, 12354–12357 CrossRef CAS PubMed.
- (a) M. Girard, D. Kindack, B. A. Dawson, J. C. Ethier and D. V. C. Awang, J. Nat. Prod., 1988, 51, 1023–1024 CrossRef CAS PubMed; (b) V. K. Tandon, D. B. Yadav, R. V. Singh, A. K. Chaturvedi and P. K. Shukla, Bioorg. Med. Chem. Lett., 2005, 15, 5324–5328 CrossRef CAS PubMed; (c) I. Gomez-Monterrey, G. Santelli, P. Campiglia, D. Califano, F. Falasconi, C. Pisano, L. Vesci, T. Lama, P. Grieco and E. Novellino, J. Med. Chem., 2005, 48, 1152–1157 CrossRef CAS PubMed; (d) A. Ventura Pinto and S. Lisboa de Castro, Molecules, 2009, 14, 4570–4590 CrossRef CAS PubMed.
- Y. Zhao, S. Yu, W. Sun, L. Liu, J. Lu, D. McEachern, S. Shargary, D. Bernard, X. Li, T. Zhao, P. Zou, D. Sun and S. Wang, J. Med. Chem., 2013, 56, 5553–5561 CrossRef CAS PubMed.
- K. K. C. Liu, J. Li and S. Sakya, Mini-Rev. Med. Chem., 2004, 4, 1105–1125 CrossRef CAS PubMed.
- S. Gafner, J.-L. Wolfender, M. Nianga, H. Stoeckli-Evans and K. Hostetmann, Phytochemistry, 1996, 42, 1315–1320 CrossRef CAS PubMed.
- C. A. Camara, T. M. S. Silva, T. G. Da-Silva, R. M. Martins, T. P. Barbosa, A. C. Pinto and M. D. Vargas, An. Acad. Bras. Cienc., 2008, 80, 329–334 CrossRef CAS PubMed.
- P. D. Mishra, S. A. Verekar, A. Kulkarni-Almeida, S. K. Roy, S. Jain, A. Balakrishnan, R. Vishwakarma and S. K. Deshmukh, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2013, 52, 565–567 Search PubMed.
- H. Hussain, K. Krohn, V. U. Ahmad, G. A. Miana and I. R. Green, ARKIVOC, 2007, 145–171 CAS.
- M. J. Teixeira, Y. M. de Almeida, J. R. Viana, J. G. Holanda Filha, T. P. Rodrigues, J. Romulo, C. Jr. Prata, I. C. B. Coelho, V. S. Rao and M. M. L. Pompeu, Phytother. Res., 2001, 15, 44–48 CrossRef CAS PubMed.
- (a) J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416–14417 CrossRef CAS PubMed; (b) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS PubMed; (c) K. Albertshofer, K. E. Anderson and C. F. Barbas, Org. Lett., 2012, 14, 5968–5971 CrossRef CAS PubMed; (d) M. d. M. Sanchez Duque, O. Basle, Y. Genisson, J.-C. Plaquevent, X. Bugaut, T. Constantieux and J. Rodriguez, Angew. Chem., Int. Ed., 2013, 52, 14143–14146 CrossRef CAS PubMed; (e) J. H. Lee and D. Y. Kim, Bull. Korean Chem. Soc., 2013, 34, 1619–1620 CrossRef CAS; (f) Y. Wang, F. Shi, X.-X. Yao, M. Sun, L. Dong and S.-J. Tu, Chem.–Eur. J., 2014, 20, 15047–15052 CrossRef CAS PubMed; (g) S. Jayakumar, S. Muthusamy, M. Prakash and V. Kesavan, Eur. J. Org. Chem., 2014, 1893–1898 CrossRef CAS; (h) C. E. T. Mitchell, S. E. Brenner and S. V. Ley, Chem. Commun., 2005, 5346–5348 RSC; (i) M. Rueping, E. Merino and E. Sugiono, Adv. Synth. Catal., 2008, 350, 2127–2131 CrossRef CAS; (j) W. Yang and D.-M. Du, Adv. Synth. Catal., 2011, 353, 1241–1246 CrossRef CAS; (k) N. Molleti and V. K. Singh, Org. Biomol. Chem., 2015, 13, 5243–5254 RSC.
- (a) B. Tan, N. R. Candeias and C. F. 3rd Barbas, Nat. Chem., 2011, 3, 473–477 CAS; (b) Y. Gao, Q. Ren, S.-M. Ang and J. Wang, Org. Biomol. Chem., 2011, 9, 3691–3697 RSC; (c) F. Zhou, M. Ding, Y.-L. Liu, C.-H. Wang, C.-B. Ji, Y.-Y. Zhang and J. Zhou, Adv. Synth. Catal., 2011, 353, 2945–2952 CrossRef CAS; (d) C. Curti, G. Rassu, V. Zambrano, L. Pinna, G. Pelosi, A. Sartori, L. Battistini, F. Zanardi and G. Casiraghi, Angew. Chem., Int. Ed., 2012, 51, 6200–6204 CrossRef CAS PubMed; (e) W. Sun, G. Zhu, C. Wu, G. Li, L. Hong and R. Wang, Angew. Chem., Int. Ed., 2013, 52, 8633–8637 CrossRef CAS PubMed; (f) Y. Shi, A. Lin, H. Mao, Z. Mao, W. Li, H. Hu, C. Zhu and Y. Cheng, Chem.–Eur. J., 2013, 19, 1914–1918 CrossRef CAS PubMed; (g) Z.-K. Fu, J.-Y. Pan, D.-C. Xu and J.-W. Xie, RSC Adv., 2014, 4, 51548–51557 RSC; (h) M. Montesinos-Magraner, C. Vila, R. Canton, G. Blay, I. Fernandez, M. C. Munoz and J. R. Pedro, Angew. Chem., Int. Ed., 2015, 54, 1–6 CrossRef PubMed.
- (a) P. Vinayagam, M. Vishwanath and V. Kesavan, Tetrahedron: Asymmetry, 2014, 25, 1252 CrossRef CAS; (b) V. Pratap Reddy Gajulapalli, P. Vinayagam and V. Kesavan, Org. Biomol. Chem., 2014, 12, 4186–4191 RSC; (c) V. P. Reddy Gajulapalli, P. Vinayagam and V. Kesavan, RSC Adv., 2015, 5, 7370–7379 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1021301. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra25025k |
| This journal is © The Royal Society of Chemistry 2016 |