
A clean synthesis approach to biocompatible amphiphilic conetworks via reversible addition–fragmentation chain transfer polymerization and thiol–ene chemistry
Abstract
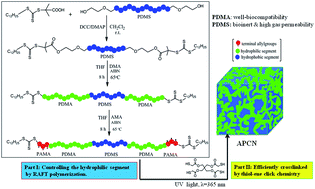
A series of amphiphilic block copolymers containing hydrophobic polydimethylsiloxane (PDMS) segments and hydrophilic poly(N,N-dimethylacrylamide) (PDMAAm) segments have been synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization, which were then crosslinked into well-defined amphiphilic conetworks (APCNs) via ultraviolet (UV) induced thiol–ene click chemistry. Briefly, a PDMS-based RAFT agent was synthesized from the esterification of trithiocarbonate and bis(hydroxyethyloxypropyl) PDMS, and was used to control the RAFT polymerization of monomer DMAAm and allyl methacrylate (AMA) to form amphiphilic copolymers with a well-defined molecular mass and narrow dispersity. The amphiphilic copolymers were then crosslinked via UV induced thiol–ene click chemistry into APCNs, which showed unique amphiphilic characteristics as well as good mechanical properties, making them potential candidates in biomaterials. Transmission electron microscopy (TEM) and atomic force microscopy (AFM) inferred that the resultant APCN exhibited the behavior of microphase separation with a small channel size and uniform phase domain. Therefore, this kind of APCN possessed excellent comprehensive properties, i.e. a well-defined and co-continuous microstructure and high water uptake properties with a homogeneous hydrophilic channel, low cytotoxicity, high mechanical strength (2.1 ± 0.7 MPa) and elongation ratio (173 ± 17%), suggesting a promising biomaterial candidate for contact lenses, drug controlled systems, biomedical scaffolds for tissue engineering and supports for biocatalysts.
 Please wait while we load your content...
Please wait while we load your content...

