DOI:
10.1039/C5RA24913A
(Paper)
RSC Adv., 2016,
6, 5278-5287
Polyphenols from pinecones of Pinus koraiensis induce apoptosis in colon cancer cells through the activation of caspase in vitro
Received
24th November 2015
, Accepted 23rd December 2015
First published on 5th January 2016
Abstract
We had previously extracted and purified a polyphenol from P. koraiensis pinecone (PPP), and evaluated its antiproliferative activities against different cancer cells lines. In the present study, we further improved the purity of PPP through the gradient elution method by different concentrations of ethanol (20%, 40% and 60%), so as to study their antitumor effects. Then, the purity and the influence upon cell viability of the purified components from PPP (PPP-20, PPP-40 and PPP-60) were evaluated. The results showed that PPP-40 had the highest phenolic purity (57.25 ± 1.83%) and exhibited the strongest inhibition (EC50 0.21 ± 0.03 mg mL−1) against LOVO cells in a dose-dependent manner. Catechin and taxifolin, the main components of PPP-40, may contribute to its antitumor activity. Moreover, we further investigated the molecular mechanisms of the PPP-40-induced apoptosis. The DNA damage in LOVO cells was observed by PI staining and comet assay. Besides, the apoptosis rates were further detected by flow cytometry. Consequently, the data showed that PPP-40 could significantly disrupt the mitochondrial membrane potential (MMP), reduce the content of adenosine 5′-triphosphate (ATP) and increase the production of reactive oxygen species (ROS). All the results indicated the mitochondrial dysfunction was involved in the PPP-40-induced apoptosis. Meanwhile, the typical markers of apoptosis involving the intrinsic and extrinsic pathways were analyzed as well. This showed that PPP-40 could not only promote the intrinsic apoptosis by increasing the release of cytochrome c (cyt c) and activating caspase-9 and -3, but also induce extrinsic apoptosis by activating caspase-8.
1. Introduction
Colon cancer is the third most common cancer in the world,1 and its incidence is increasing annually.2 Despite of the rapid development of various targeted therapeutic approaches, mortality related to colon cancer remains at a staggering high level.3 Therefore, more effective therapeutic strategies are put forward to combat colon cancer. In this regard, it is increasingly accepted that natural polyphenols can be used as an alternative therapy because they generally have high therapeutic effectiveness as well as low side effects.4,5 The possible antitumor mechanisms of polyphenols include interference with many intracellular signaling pathways that regulate cell survival or apoptosis.6–8
The basic mechanisms of apoptosis have been divided depending on the trigger of the death programs.9 In mammalian cells, apoptosis may be activated by two ways, intrinsic (mitochondrial-mediated)10 and extrinsic (death receptor-mediated)11,12 pathways which both activate apoptotic effector molecules to induce apoptosis.13,14 Differently, the intrinsic pathway is activated by the release of pro-apoptotic factors such as cyt c from the mitochondrial matrix following the loss of inner mitochondrial membrane integrity and activation of caspase-9. It's in accordance with report of Roy et al. that the polyphenols of black tea induce the apoptosis of skin cancer cells by the intrinsic pathway.15 While the extrinsic pathway is initiated by the binding of extracellular death ligands to their cell-surface death receptors which leads to the activation of caspase-8, and then the apoptosis of cells.16,17 Similar research could be found that mycelia and culture supernatants of Cordyceps militaris induced the apoptosis in human glioblastoma GBM8401 cells through activation of caspase-3 and caspase-8.18
In the last few years, a wide range of biological activities of Pinaceae extracts, especially pine polyphenols, have been reported.19,20 Pinup koraiensis (P. koraiensis), a member of Pinaceae plants, also possesses a variety of biological utilizing potentials.21 However, P. koraiensis pinecone, the by-product in processing of seeds, is commonly burnt as firewood or discarded as waste, which leads to the waste of biological resources. Our laboratory has demonstrated that P. koraiensis pinecones contain plenty of biological active substances, among which polyphenols are the most important components.22,23 Researches on the pharmacological activity and applications in medical science of pinecones have been reviewed. For example, the studies of Pinaceae have found that the pinecones extracts, like pine barks extracts, possess potent antioxidant activity.22,24 However, the relevant research in its effect upon colon cancer had not been reported. Recently, our previous study had revealed that the purified polyphenols from P. koraiensis pinecones could significantly inhibit the proliferation of LOVO cells.23 In the present study, we sought to detect the antitumor effects of the purified polyphenols from pinecones of P. koraiensis on LOVO cells and study their antitumor mechanisms, which may involve the apoptosis associated with activation of caspases.
2. Materials and methods
2.1 Materials
P. koraiensis pinecones were provided by Yichun Hongxing District Forestry Bureau (Yichun, China). HPLC-grade catechin, epicatechin, procyanidin B2 and taxifolin were purchased from Shanghai Yuanye Biological Technology Co., Ltd. (Shanghai, China). The Roswell Park Memorial Institute 1640 (RPMI-1640) and Fetal Bovine Serum (FBS) were provided from Hyclone laboratory (South Logan, Utah, USA). MTT, RNase A and PI were purchased from Sigma-Aldrich Chemicals (St. Louis, MO, USA). Annexin V-FITC/PI and ROS test kits were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China), Caspase-3, -8, -9 and z-VAD-fmk (Z-VAD) were purchased from Beyotime Company (Shanghai, China). All other chemicals were of analytical grade and purchased from local suppliers.
2.2 Preparation of samples
We previously extracted and purified a polyphenol from P. koraiensis pinecones (PPP) with the purity of 27.93 ± 0.14%.23 In order to obtain higher purity, the gradient elution of PPP was carried out in glass columns packed with pretreated D101 macroporous resins.25 The concentrations of ethanol–water in the gradient elution were 20%, 40% and 60% (v/v), respectively. Operating conditions were as follows: flow rate, 2.0 mL min−1; collected volume, 2 bed volumes for each gradient. Finally, the collected components (PPP-20, PPP-40 and PPP-60) were concentrated by a rotary evaporator to remove the ethanol in solution. Then the purities of polyphenols were measured according to the method described in our previous study.23
2.3 Cell viability assay
Human colon cancer cells line (LOVO) was gifted by the School of Life Science of Harbin Institute of Technology. The effect of PPP-40 on the viability of LOVO cells was evaluated by the MTT assay.26–28 LOVO cells (1 × 105 cells per mL) were transferred to 96-well plates containing RPMI-1640 medium. After 12 h, the medium was changed to 100 μL of growth medium containing various concentrations of samples in each well and incubated for another 44 h at 37 °C in 5% CO2. Then, MTT solution (5 mg mL−1) was added to each well. After 4 h of incubation, the precipitated formazan was dissolved in 150 μL DMSO, and the absorbance was read at 490 nm using a microplate reader (Model 680, Bio-Rad, Hercules, CA, USA).
2.4 Toxicity of splenocytes
Briefly, spleens collected from the rats under aseptic conditions were chopped into small pieces and passed through a mesh screen to obtain a homogeneous cell suspension. The splenocytes were harvested by centrifugation (1000 × g for 10 min), the red blood cells were lysed by the lysis buffer. Recovered cells were washed twice and adjusted to 1 × 106 cells per mL. The purified cells were used directly for the cytotoxicity assay by MTT method.26–28
2.5 HPLC analysis
PPP-40 was analyzed by a HPLC (Agilent 1260 Series, Agilent Technologies, USA) equipped with an Eclipse XDB-C18 column (5 μm, 250 mm × 4.6 mm, Agilent). Eluent (A) and eluent (B) were 1.0% acetic acid and methanol, respectively. The mobile phase conditions: 0–40 min, 95A/5B – 70A/30B; 40–65 min, 70A/30B – 50A/50B; 65–66 min, 50A/50B – 95A/5B; 66–75 min, 95A/5B. The operating conditions were: detection wavelength, 280 nm; flow rate, 0.6 mL min−1; injection volume, 10 μL.
2.6 Colony formation assay
LOVO cells from different groups (0, 50, 100, 200, 300 and 400 μg mL−1 of PPP-40) were seeded in 6-well plates with a density of 300 cells per well for 14 days. At the end of the experiment, the medium was removed and the remaining cells were washed twice with PBS. Colonies were fixed in methanol for 20 min and then stained with 0.25% gentian violet solution. The plates were washed gently and then scanned by a fluorescence microscope (Olympus, C5060-ADU, Tokyo, Japan). The number of colonies containing at least 50 cells was counted and colony forming efficiency was calculated (percentage of colonies = number of colonies formed/number of cells inoculated × 100%).
2.7 PI staining
LOVO cells were treated with 200 μg mL−1 PPP-40 for 24 h, harvested at a concentration of 1 × 106 cells per mL, washed with PBS twice, and then resuspended in 160 μL PBS. Finally, the cells were stained with 50 μg mL−1 PI for 15 min at 25 °C in the dark. After centrifugation, the pellets were dispersed in PBS. The specimens were examined under a fluorescence microscope (Olympus, C5060-ADU, Tokyo, Japan).
2.8 Measurement of DNA damage by comet assay
LOVO cells (1 × 106 cells per mL) were incubated with 0, 100, 200 and 400 μg mL−1 PPP-40 for 48 h and then harvested with 0.25% trypsinization for 1–2 min. Subsequently, the comet assay was performed as described by Liu et al.29 Briefly, the cells were collected, rinsed and suspended in PBS. Cells suspension (30 μL, 1 × 104 cells) was mixed with 100 μL 1% (v/v) molten low-melting-point agarose at 37 °C. The mono-suspension was cast on a microscopic slide covered with a layer of 0.8% regular-melting-point agarose. The agarose was gelled at 4 °C for 10 min and the slide was then immersed in a cold, freshly prepared lysing solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris–HCl, 1% Triton-X 100, 10% DMSO, pH 10) for 30 min at 4 °C. After lysis, the slides were washed three times by distilled water and immersed in fresh alkaline electrophoresis solution containing 300 mM NaOH, 1 mM EDTA (pH 13) for 10 min at 4 °C. Electrophoresis was carried out in ice bath (4 °C) at 300 mA and 25 V for 20 min. The slides were neutralized to pH 7.5 with a 0.4 M Tris buffer, and stained with 50 μL 20 μg mL−1 ethidium bromide solution in the dark for 20 min. The slides were washed in chilled distilled water for 10 min to neutralize the excess alkali, air-dried and then screened for comets by fluorescence microscopy (Olympus, C5060-ADU, Tokyo, Japan) with a 515–560 nm excitation filter and a 590 nm barrier filter.
2.9 Apoptosis measurement by flow cytometry
LOVO cells (1 × 106 cells per well) were seeded in 6-well plates for 12 h. Then, the cells were treated with 0, 100, 200 and 400 μg mL−1 PPP-40 for 24 and 48 h, collected, washed, and then resuspended in 500 μL of binding buffer containing 5 μL Annexin V-FITC and 5 μL PI. The cells were incubated in the dark for 10 min at 25 °C, and then analyzed by the flow cytometer (FCM) (BD Biosciences, San Jose, CA, USA).30
2.10 Mitochondrial membrane potential (MMP) detection
Briefly, LOVO cells (1 × 106 cells per well) were seeded in 6-well plates for 12 h. Then, the cells were treated with 0, 100, 200 and 400 μg mL−1 PPP-40. After the indicated treatments for 48 h, cells were incubated with JC-1 staining solution (5 μg mL−1) for 20 min at 37 °C, rinsed twice with PBS and then imaged under a fluorescence microscope (Olympus, C5060-ADU, Tokyo, Japan) with excitation 488 nm/emission 535 nm for JC-1 green and excitation 488 nm/emission 595 nm for JC-1 red.
2.11 Determination of ATP content
ATP content was measured by the ATP Determination Kit (Invitrogen Corp) following the instructions of the manufacturer. Briefly, LOVO cells (1 × 106 cells per mL) were washed twice by PBS, lysed in a lysis buffer and then centrifuged at 1000 × g for 10 min. The supernatant was mixed with the ATP Determination Kit reaction buffer. After mixing gently, cellular ATP level could be calculated and expressed as a percentage of the control group.
2.12 Reactive oxygen species (ROS) detection
LOVO cells (1 × 106 cells per well) were treated with PPP-40 at 0, 100, 200 and 400 μg mL−1 for 48 h, respectively. Afterwards, the cells were stained with 10 μM DCFH-DA for 30 min subsequently, the cells were collected, resuspended in PBS. Finally, fluorescence was imaged with fluorescent microscope at an excitation wavelength of 488 nm and emission of 525 nm (Olympus, C5060-ADU, Tokyo, Japan).
2.13 Determination of cyt c
LOVO cells were treated with various concentrations of PPP-40 for 48 h. Subsequently, the cells were extracted by lysis buffer from cyt c ELISA kit (Enzo Life Sciences). Briefly, the cells were collected after centrifugation, resuspended with cell permeabilization buffer, then vortexed, incubated on ice for 5 min, and finally centrifuged. The supernatants containing the cytosolic fraction of cyt c were saved. The remaining pellets were resuspended with RIPA, cell lysis buffer 2, vortexed and incubated on ice for 5 min again. The lysate was vortexed and centrifuged. Similarly, the supernatants containing the mitochondrial fraction of cyt c were saved. The optical density of each fraction was measured at 405 nm by a microplate reader (Model 680, Bio-Rad, Hercules, CA, USA).
2.14 Determination of caspase-3, -8 and -9 enzymatic activities
LOVO cells (1 × 106 cells per well) were seeded in 6-well plates and incubated at 37 °C for 48 h with various concentrations of PPP-40 pretreatment. At the end of the experiments, the cells were harvested by trypsinization and lysed in a lysis buffer for 30 min on ice. The cells were then washed with ice-cold PBS and centrifuged (1000 × g for 5 min). The cell pellets were resuspended in 100 μL of ice-cold assay buffer. Under the condition of strong vortex mixing, the cells were lysed and then the suspension was used for assays according to the manufacturer's protocols. Briefly, 10 μL of the cell lysates was incubated with 80 μL of the assay buffer and 10 μL of the substrate (caspase-3; Ac-DEVD-pNA, caspase-8; Ac-IETD-pNA, and caspase-9; Ac-LEHD-pNA, respectively) at 37 °C for 2 h. Finally, the release of p-nitroaniline (pNA) was measured at 405 nm by a microplate reader (Model 680, Bio-Rad, Hercules, CA, USA). Results can be represented as the multiples of increase in enzyme activities compared with the untreated control.
2.15 Statistical analysis
Data were expressed as means ± standard deviation (SD) of three independent measurements. All the values were expressed as means ± SD. Statistical analysis was performed by Student t-test and one-way ANOVA by SPSS (Version 18.0). Differences at p < 0.05 were considered statically significant.
3. Results
3.1 Purification of the different gradients of PPP
Ethanol solution was adopted as the eluent, which could effectively desorb polyphenols from resin.31 The concentration of ethanol significantly affected the phenolic purity.31,32 In order to obtain higher purity of polyphenols, the gradient elution of ethanol was performed by 20, 40 and 60% (v/v) ethanol–water solution. As seen in Table 1, three purified components of PPP (PPP-20, PPP-40, PPP-60) were obtained with the purity of 18.25 ± 2.52%, 57.25 ± 1.83%, 31.32 ± 3.38%, respectively. Obviously, PPP-40 had the highest phenolic purity, which increased to 2.05 times that of PPP. The results indicated that the 40% was the most suitable ethanol concentration for the purification of PPP, which may be concerned with the polarity of solvent and the solubility of PPP.25
Table 1 Phenolic purity of the different gradients of PPP (n = 3)
| Samples |
Phenolic purity (%) |
| PPP-20 |
18.25 ± 2.52 |
| PPP-40 |
57.25 ± 1.83 |
| PPP-60 |
31.32 ± 3.38 |
3.2 Cytotoxicity of PPP-40
As shown in Fig. 1A, the samples reduced the viability of LOVO cells after 48 h treatments in dose-dependent manner, of which PPP-40 showed the highest phenolic purity (Table 1) and the strongest effect upon the viability of LOVO cells, declining from 87.98 ± 2.54% to 19.61 ± 2.91%. For the rest samples, PPP-20 displayed relatively poorer viability (47.24 ± 2.02%) and lower phenolic purity (18.25 ± 2.52%). Concentration of PPP-40 which reduced 50% of LOVO cells viability (EC50) was the lowest (0.21 ± 0.03 mg mL−1), whereas the EC50 of PPP was 0.36 ± 0.03 mg mL−1 (Fig. 1B), which indicated the further purification could provide an enhanced antitumor effect. Fig. 1C also showed that further purification produced better results in immunoregulatory activity. As a result, the fact that PPP-40 was able to reduce the viability of cancer cells (LOVO cells) selectively, but not normal cells (splenocytes), strongly nominated PPP-40 as a chemotherapeutic candidate.
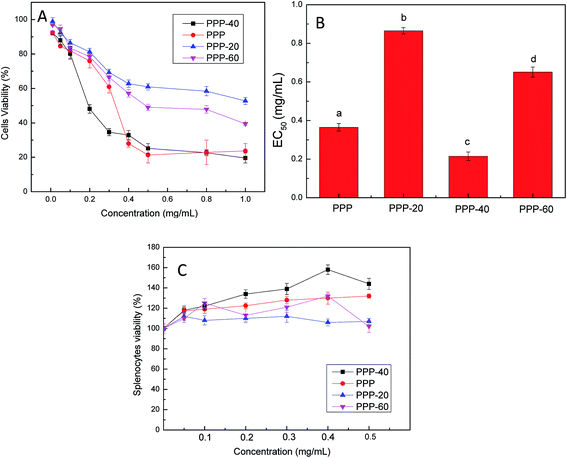 |
| | Fig. 1 Effects of the purified PPP on cells viability. (A) Effects of LOVO cells viability with treatment of different purified components of PPP. (B) EC50 values for the inhibition of LOVO cells viability by the different purified components of PPP. (C) Splenocyte cytotoxicity of different purified components of PPP. Results were represented as means ± standard deviation of three parallel measurements. Bars with no letters in common were significantly different (p < 0.05). | |
3.3 HPLC analysis of PPP-40
By comparing the retention time of standards in HPLC chromatogram (Fig. 2A), the compositions of PPP-40 were evaluated. As seen in Fig. 2B, PPP-40, containing mainly catechin (205.98 ± 4.11 μg mL−1) and taxifolin (12.16 ± 1.02 μg mL−1), reduced the viability of LOVO cells significantly. Similarly, Annurca apple peel with higher amounts of procyanidins possessed more effective antitumor function.33 Araujo et al. also reported the antitumor effects of dietary polyphenols including catechin in colon cancer cell lines.34
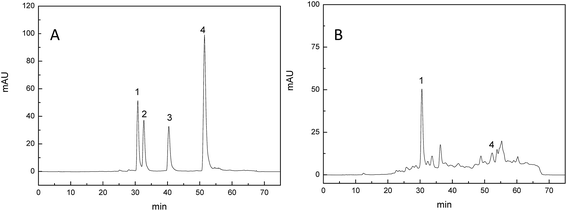 |
| | Fig. 2 HPLC chromatograms of standard (A) and PPP-40 (B) recorded at 280 nm (peak 1: catechinic acid, peak 2: procyanidin B2, peak 3: epicatechin, peak 4: taxifolin). | |
3.4 Effect of PPP-40 on colony formation in LOVO cells
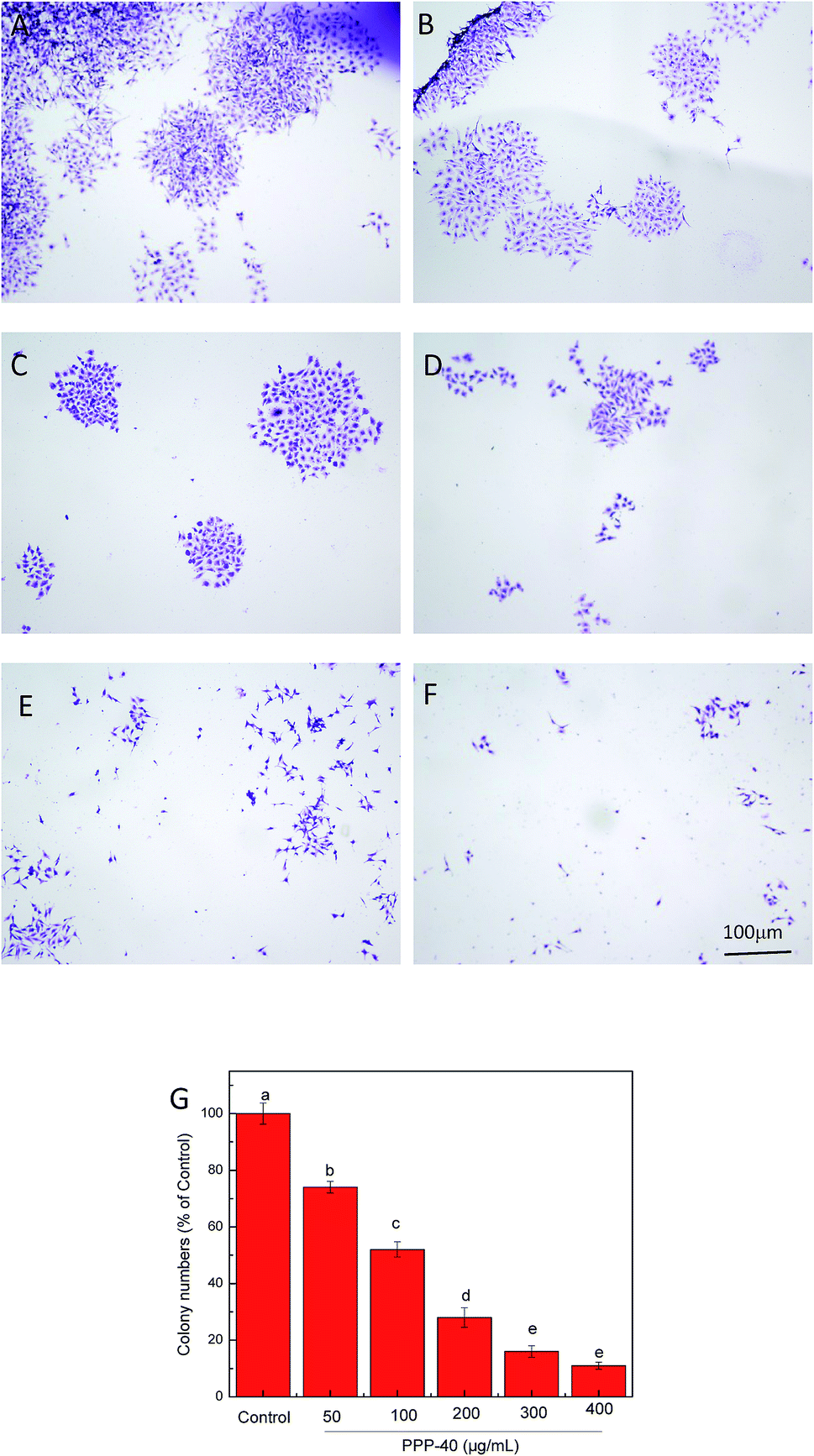
As shown in Fig. 3, PPP-40 significantly diminished the capacity of LOVO cells to form colonies. Cells were treated with PPP-40 at 50, 100, 200, 300 and 400 μg mL−1, while the colony forming efficiency were 74.12 ± 2.07%, 52.02 ± 2.48%, 28.35 ± 3.54%, 16.07 ± 2.07% and 11.12 ± 1.23%, respectively. Compared with control group, the 400 μg mL−1 PPP-40 induced the reduction in the number of colonies by about 90% (p < 0.05). Similarly, the apple polyphenol could also significantly suppress colony formation of DLD-1 cells.35
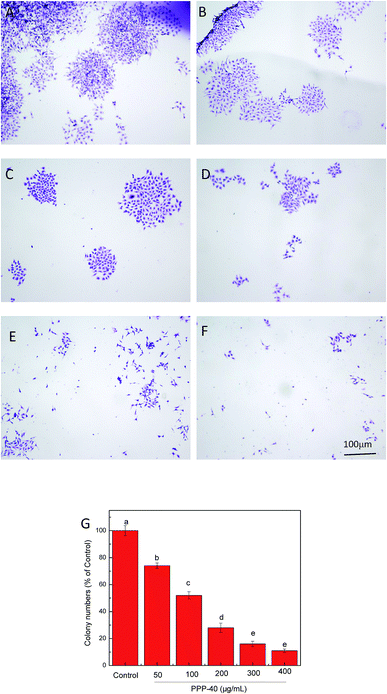 |
| | Fig. 3 PPP-40 prevented colony formation assay. Colony formation inhibitions of PPP-40 at 0 (A), 50 (B), 100 (C), 200 (D), 300 (E) and 400 μg mL−1 (F) on LOVO cells were examined by fluorescence microscopy (magnification, 100×). Colony formation inhibition rates of PPP-40 on LOVO cells (G). Results were represented as means ± standard deviation of three parallel measurements. Bars with no letters in common were significantly different (p < 0.05). | |
3.5 Screening of PPP-40 for DNA damage
Based on the results above, we further explored the effects on the apoptosis of LOVO cells. Morphological changes of nuclei in LOVO cells, which were treated with or without 200 μg mL−1 PPP-40, were confirmed by PI staining (Fig. 4). Compared with the round and homogeneously stained nuclei in the control group (Fig. 4A), the nuclear shrinkage and disappearance were observed following PPP-40 treatment and shown in Fig. 4B by arrows, which were both typical characteristics of apoptosis. DNA damage is a well-recognized hallmark of apoptosis.36 Comet assay could rapidly detect the level of DNA damage as well.37,38 As shown in Fig. 5A, the untreated LOVO cells had the comet heads with high-density of DNA accompanied with smooth margins and intact nuclei. After treatment of PPP-40, the clear and comet-like tails in the migration of DNA could be observed (Fig. 5B–D). These results suggested that PPP-40 could induce DNA damage in LOVO cells, which would further lead to apoptosis.
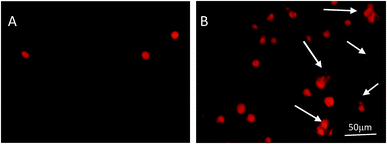 |
| | Fig. 4 The assessment of nuclear morphology of LOVO cells by PI staining. LOVO cells treated with 200 μg mL−1 PPP-40 for 24 h were stained with PI. Untreated cells were used as control (A), the cells treated with 200 μg mL−1 PPP-40 (B). The results were examined by fluorescence microscopy (magnification, 200×). Nuclear shrinkage, irregular nuclei and nuclei disappearance were shown by arrows. | |
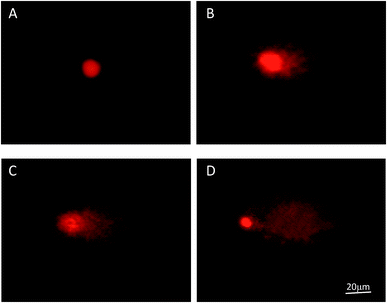 |
| | Fig. 5 Typical fluorescence images of comet of LOVO cells after incubating with PPP-40 (magnification, 400×): 100 (B), 200 (C) and 400 μg mL−1 (D) for 48 h. Untreated cells were used as control (A). | |
3.6 Effect of PPP-40 upon the apoptosis of LOVO cells
Levels of PPP-40-induced apoptosis (the early and late apoptosis rate) were quantified by FACS analysis after staining with Annexin V/PI. As shown in Fig. 6, the amount of apoptotic cells increased obviously following the PPP-40 treatment in a dose- and time-dependent manner. Especially, as shown in Fig. 6h, cells treated with 400 μg mL−1 PPP-40 for 48 h presented obvious apoptosis (41.50 ± 1.38%) than cells in control (1.31 ± 0.55%). Some other rates of apoptosis were shown in Fig. 6C.
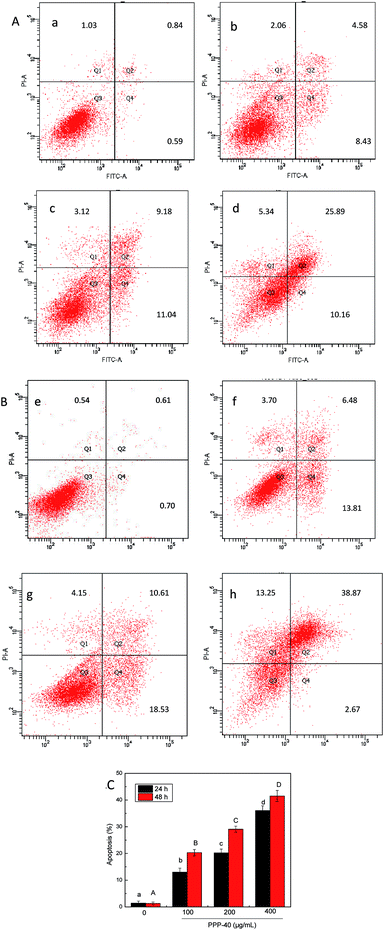 |
| | Fig. 6 Effects of PPP-40 on LOVO cells apoptosis by flow cytometry analysis using the Annexin V-FITC/PI double staining assay. (A) Cells were treated with PPP-40 at 0 (a), 100 (b), 200 (c) and 400 μg mL−1 (d) for 24 h, respectively. (B) Cells were treated with PPP-40 at 0 (e), 100 (f), 200 (g) and 400 μg mL−1 (h) for 48 h, respectively. Numbers are representative of three independent experiments and indicate the percentage of cells in each quadrant. (C) The percentage of apoptotic cells (early and late apoptotic cells) of PPP-40 on LOVO cells. Column chart showed mean values of three experiments. Results were represented as means ± standard deviation of three parallel measurements. Bars with no letters in common were significantly different (p < 0.05). | |
3.7 Effect of PPP-40 on the MMP in LOVO cells
Depletion of MMP in response to various stimulus results in activation of caspases and initiation of apoptotic cascades.39 Therefore, we investigated if PPP-40-induced apoptosis was associated with the depletion of MMP. MMP was examined by observing the relative ratio of red to green fluorescence of JC-1. As shown in Fig. 7A, it was found that the cells in the control group (Fig. 7A) emitted red fluorescence, suggesting the control cells had a normal MMP. However, as shown in Fig. 7B–D, treatment with increasing concentrations of PPP-40 for 48 h led to a gradually larger population of cells in depolarized MMP, which were confirmed by the increase of green fluorescence and decrease of red fluorescence.
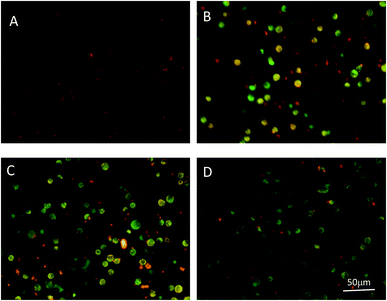 |
| | Fig. 7 PPP-40 induced MMP collapse in LOVO cells. Cells were treated with PPP-40 at 100 (B), 200 (C), and 400 μg mL−1 (D) for 48 h. Untreated cells were used as control (A). JC-1 fluorescence were visualized using the fluorescence microscope (magnification, 200×). | |
3.8 Reduction of ATP in LOVO cells by PPP-40
MMP is the driving force for ATP synthesis. Loss of MMP is expected to cause decreases in ATP levels in cells. Therefore, the levels of cellular ATP were determined after PPP-40 treatment. As shown in Fig. 8, treatment with increasing concentrations of PPP-40 in LOVO cells for 48 h significantly reduced the ATP concentration to 77.34 ± 2.46%, 64.25 ± 2.31% and 39.17 ± 3.75% compared with that of control cells, respectively.
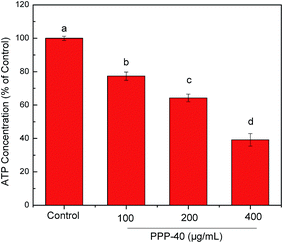 |
| | Fig. 8 Effects of PPP-40 on the ATP level of LOVO cells. The results were expressed as a percentage of the control, which was set at 100%. Results were represented as means ± standard deviation of three parallel measurements. Bars with no letters in common were significantly different (p < 0.05). | |
3.9 Effect of PPP-40 on the production of ROS
The mitochondrial apoptosis pathway can be triggered by many stimuli, including ROS. Mitochondria are the major site of ROS production, and the accumulation of ROS may lead to the initiation of apoptosis.40,41 Therefore, we analyzed the production of intracellular ROS in PPP-40-treated cells by using a fluorescent probe DCFH-DA. As shown in Fig. 9, in the control group, no bright fluorescence image was found. However, the brighter fluorescence images were detected after treatment with different concentrations of PPP-40 for 48 h, which indicated that ROS production obviously increased in dose-dependent way.
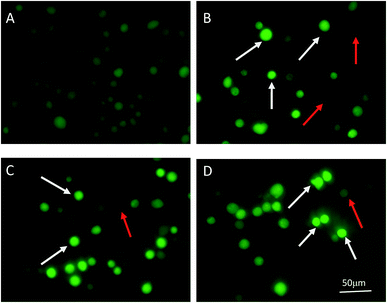 |
| | Fig. 9 ROS generation level in the LOVO cells incubated with PPP-40 at 0 (A), 100 (B), 200 (C), and 400 μg mL−1 (D) for 48 h was detected by examining the fluorescence of DCF in cells by the fluorescence microscope (magnification, 200×). The enhanced ROS production was marked with emitting brighter DCF fluorescence and shown by the white arrow, no expression ROS was dark fluorescent image and shown by the red arrow. | |
3.10 Release of cyt c by PPP-40 in LOVO cells
Generally, the dissipation of MMP induces the release of apoptogenic factors such as cyt c from mitochondria to the cytosol.42 As shown in Fig. 10, the results showed that cyt c gradually accumulated in the cytosol, whereas the levels of cyt c in the mitochondria decreased after treatment with PPP-40 progressively. These results showed that the intrinsic apoptosis pathway was involved in the PPP-40-induced apoptosis.
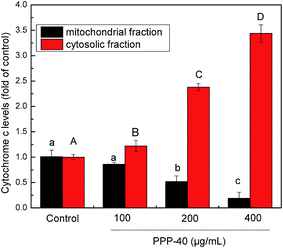 |
| | Fig. 10 Cytochrome c levels in both mitochondrial and cytosolic fraction were measured using cytochrome c ELISA kit. Results were represented as means ± standard deviation of three parallel measurements. Bars with no letters in common were significantly different (p < 0.05). | |
3.11 Activation of caspase by PPP-40 in LOVO cells
Targeting apoptotic pathways in premalignant and malignant cells is an effective strategy for cancer prevention and treatment.43 Caspase protease family is believed to play a central role in mediating various apoptotic responses.44 In the present study, we determined the activities of caspase-3, -8 and -9 via measurement of their active forms, respectively. As represented in Fig. 11, PPP-40 treatment significantly improved the activities of caspase-3, -8 and -9 in LOVO cells in dose-dependent way. Compared with untreated cells, the activity of caspase-8 in the LOVO cells treated with PPP-40 at 100, 200 and 400 μg mL−1 increased by 1.66 ± 0.09 (p < 0.05), 2.45 ± 0.17 (p < 0.05) and 2.97 ± 0.07 fold (p < 0.01), respectively, while the activity of caspase-9 increased by the 1.89 ± 0.14 (p < 0.05), 2.34 ± 0.08 (p < 0.05) and 3.23 ± 019 (p < 0.01), respectively. Similarly, the caspase-3 activity enhanced following the treatment of PPP-40 as well. These results indicated that PPP-40 resulted in activation of caspases and initiation of apoptotic cascades. To confirm caspase activation's role in PPP-40-induced apoptosis, cells were pre-incubated with a pan-caspase inhibitor, z-VAD-fmk (40.0 μM), for 2 h and then treated with 200 μg mL−1 PPP-40, inhibiting the activation of caspases completely and causing a partial reduction of cell viability compared with the one treated by PPP-40 alone (Fig. 12).
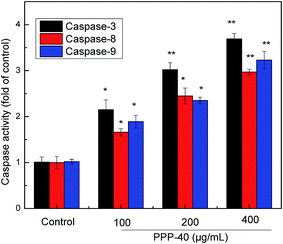 |
| | Fig. 11 Effects of PPP-40 on activation of caspases. After LOVO cells were treated with PPP-40 (100, 200 and 400 μg mL−1) for 48 h respectively, the activities of caspase-3, -8 and -9 were detected by Caspase Determination Kit. Results were represented as means ± standard deviation of three parallel measurements. *p < 0.05, **p < 0.01 vs. control group. | |
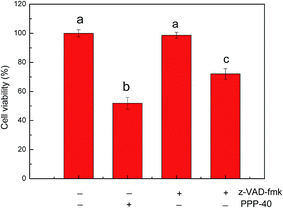 |
| | Fig. 12 Effects of caspases on PPP-40 induced apoptosis. Pan-caspase inhibitor, z-VAD (40 μM) was added to LOVO cells 2 h before the treatment with PPP-40 (200 μg mL−1), respectively. After 48 h, the cell viability was determined. Results were represented as means ± standard deviation of three parallel measurements. Bars with no letters in common were significantly different (p was added to LOVO cells 2 h before the treatment with PPP-40 (200 μg mL−1), respectively. After 48 h, the cell viability was determined. Results were represented as means ± standard deviation of three parallel measurements. Bars with no letters in common were significantly different (p < 0.05). | |
4. Discussion
Our previous study had firstly clarified the structural characterization and predicted the 3D model of the PPP purified from P. koraiensis pinecones.23 In the present study, PPP was further purified to obtain the PPP-40 with higher purity. The data obtained (Fig. 1A–C) was in a good agreement with our expectation that the viability of cancer cells (LOVO cells) were reduced by PPP-40 while normal cells (splenocytes) were less affected by the treatment of PPP-40. These results were consistent with the previous researches of the antitumor effects of polyphenols in colon cancer cells.45,46
Moreover, there was a considerable difference in the sensitivity of LOVO cells to different purified components from PPP, of which PPP-40 possessed the strongest antitumor activity (Table 1). However, the concentrations of samples have been normalized, which suggested that different responses on the viability of cells were associated with the phenolic purity and components, rather than the dose. In fact, PPP-40 dose contain the highest amount of polyphenols. A recent study has also reported that a phenolic fraction from mango peels contained plenty of polyphenols and exhibited good antitumor activities.47 Based on our HPLC analysis, catechin and taxifolin mainly exist as bioactive components of PPP-40 (Fig. 2). Thus, we speculated that their synergism might be involved in the apoptosis-based cytotoxicity of PPP-40 against LOVO cells. However, the antitumor effects of the individual polyphenol component present in PPP-40 are also required to be further studied.
Apoptosis plays a crucial role in the progress of proliferation, differentiation, senescence and death in cell.48 Nuclear shrinkage, irregular nuclei, chromatin condensation and DNA fragmentation are typical characteristics of apoptosis.49,50 After being treated with PPP-40, several typical characteristics of apoptosis, like cell shrinkage and DNA fragmentation, were observed and apoptosis rates were quantified by FCM. These results demonstrated the occurrence of apoptosis in PPP-40-treated LOVO cells and were in accordance with the application of other polyphenols on colon cells. For example, the crude extract of Rheum palmatum L could induce DNA damage in human colon cancer cells and promote apoptosis in dose-dependent manner.51 Further studies were performed to examine the mechanisms of apoptosis induction by PPP-40 in LOVO cells and find PPP-40-activated caspase-8, -9 and -3 (Fig. 11), which preliminary proved that both intrinsic and extrinsic pathways might be involved in PPP-40-induced cells apoptosis. However, blocking caspase activities by pretreating cells with a pan-caspase inhibitor didn't prevent completely PPP-40-induced the reduction of cell viability (Fig. 12). Therefore, the data support that PPP-40-induced apoptosis in LOVO cells could be, at least in part, dependent on caspase activation.
Disruption of the MMP usually accompanies or initiates apoptosis.39 Previous reports also showed that loss of MMP was an early event in polyphenols-induced apoptosis.52,53 In line with this fact, we found that the disruption of the MMP (Fig. 7) by PPP-40 seems to precede the release of cyt c (Fig. 10) from mitochondria, followed by the activation of caspase-9 and caspase-3. There results further confirmed that PPP-40 induced apoptosis via the intrinsic apoptotic pathway in LOVO cells. Furthermore, it has been generally agreed that mitochondrial dysfunction participated in the induction of apoptosis and even played a pivotal role in the apoptotic process.54,55 In the present study, the PPP-40-induced apoptosis is also accompanied by effects on mitochondria, including a significant disruption of the MMP, a reduction in ATP concentration (Fig. 8) and an elevation in ROS production (Fig. 9). These findings suggest that PPP-40 efficiently activates the intrinsic apoptotic pathway due to mitochondrial dysfunction and finally leads to apoptosis in LOVO cells. Considering the high metabolic rate in cancer cells, ATP depletion might impair the downstream of ATP-dependent processes, leading to cell death furtherly.56 Moreover, compared with normal cells, tumor cells, depend more on glycolysis to produce ATP. However, PPP-40 induced a dose-dependent decrease in cellular ATP levels, suggesting that PPP-40 might also disturb the metabolism by changing the level of glycolysis in LOVO cells. This possibility was supported by reports that resveratrol inhibited glycolysis through the PI3K/Akt/m TOR signaling pathway in human cancer cells.57,58
The extrinsic apoptosis pathway begins outside the cell by the binding of death-inducing ligands to their suitable death receptors on the cell surface. This is followed by receptor clustering and transmission of the intracellular signals that ultimately result in the destruction of the cell through the activation of caspase-8.16,17 Currently, we could not rule out the potential involvement of extrinsic pathway of apoptosis in PPP-40 treated cells, which is supported by PPP-40-induced activation of caspase-8.
5. Conclusions
In summary, we have firstly demonstrated that P. koraiensis pinecone contains rich polyphenols, and its purified component (PPP-40) possesses higher level of anticancer capacity against LOVO cells. Based on our study, we proposed a model by which PPP-40 induced LOVO cells (Fig. 13). It provided further evidence that the antitumor activities of PPP-40 in LOVO cells were primarily mediated by activation of caspase and dysregulation of mitochondria. Our results clearly suggested P. koraiensis pinecons could be exploited as a promising candidate for cancer prevention and treatment.
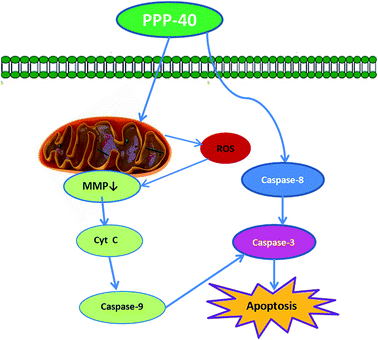 |
| | Fig. 13 Overview of pathways for PPP-40-induced apoptosis in LOVO cells. | |
Conflict of interest
The authors have declared no conflict of interest.
Nonstandard abbreviations
| P. koraiensis | Pinus koraiensis |
| PPP | Polyphenols of P. koraiensis pinecone |
| GAE | Gallic acid equivalent |
| PBS | Phosphate buffer saline |
| DMSO | Dimethylsulfoxide |
| ATP | Adenosine 5′-triphosphate |
| Cyt c | Cytochrome c |
| MMP | Mitochondrial membrane potential |
| DCFH-DA | 2′,7′-Dichlorfluorescein-diacetate |
| ROS | Reactive oxygen species |
Acknowledgements
The authors gratefully acknowledge the financial support by National Natural Science Foundation of China (no. 31170510). The authors are very grateful to Professor Lu Li guidance on language.
References
- J. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser, C. Mathers, M. Rebelo, D. M. Parkin, D. Forman and F. Bray, Int. J. Cancer, 2015, 136, 359 CrossRef PubMed.
- X. Qu, R. Xie, L. Chen, C. Feng, Y. Zhou, W. Li, H. Huang, X. Jia, J. Lv, Y. He, Y. Du, W. Li, Y. Shi and W. He, Genomics, 2013, 104, 242 CrossRef PubMed.
- J. Chai and M. M. Jamal, World J. Gastroenterol., 2012, 18, 6521 CrossRef CAS PubMed.
- G. V. Margarita, G. C. Marta, R. C. Arantxa and R. M. Ana, Nutr. Rev., 2013, 71, 585 CrossRef PubMed.
- C. Weidnera, M. Rousseaua, A. Plautha, S. J. Wowroa, C. Fischera, H. Abdel-Azizb and S. Sauera, Phytomedicine, 2015, 22, 262 CrossRef PubMed.
- A. Guzalnur, M. Fatima, H. Li, A. Guzalnur, A. Tangnur, A. Nurmuhammat and U. Halmurat, BMC Complementary Altern. Med., 2015, 15, 530 Search PubMed.
- E. Rudolf, H. Andelová and M. Cervinka, Anti-Cancer Agents Med. Chem., 2007, 7, 559 CrossRef CAS PubMed.
- M. H. Pan, C. S. Lai, J. C. Wu and C. T. Ho, Mol. Nutr. Food Res., 2011, 55, 32 CAS.
- N. Yadav and D. Chandra, Mitochondrion, 2014, 16, 18 CrossRef CAS PubMed.
- W. Pang, X. Leng, H. Lu, H. Yang, N. Song, L. Tan, Y. Jiang and C. Guo, Neurosci. Lett., 2013, 552, 140 CrossRef CAS PubMed.
- Y. J. Kang, I. Y. Kim, E. H. Kim, M. J. Yoon, S. U. Kim, T. K. Kwon and K. S. Choi, Exp. Mol. Med., 2011, 43, 24 CrossRef CAS PubMed.
- D. M. Zhu, J. Shi, S. Liu, Y. Liu and D. Zheng, PLoS One, 2011, 6, e18291 CAS.
- S. H. M. Kenzie and A. C. Clark, Curr. Cancer Drug Targets, 2008, 8, 98 CrossRef.
- D. Brenner and T. W. Mak, Curr. Opin. Cell Biol., 2009, 21, 871 CrossRef CAS PubMed.
- P. Roy, N. Nigam, J. George, S. Srivastava and Y. Shukla, Cancer Biol. Ther., 2009, 8, 1281 CrossRef CAS PubMed.
- Z. Jin and W. S. El-Deiry, Cancer Biol. Ther., 2005, 4, 139 CAS.
- P. Hensley, M. Mishra and N. Kyprianou, Biol. Chem., 2013, 394, 831 CrossRef CAS PubMed.
- C. H. Yang, Y. H. Kao, K. S. Huang, C. Y. Yang and L. W. Lin, Cell Death Dis., 2012, 3, e431 CrossRef PubMed.
- Y. J. Peng, C. H. Lee, C. C. Wang, D. M. Salter and H. S. Lee, Free Radical Biol. Med., 2012, 52, 765 CrossRef CAS PubMed.
- D. S. Kim, M. S. Kim, S. W. Kang, H. Y. Sung and Y. H. Kang, J. Agric. Food Chem., 2010, 58, 7088 CrossRef PubMed.
- X. Yang, H. Zhang, Y. Zhang, Y. Ma and J. Wang, Fitoterapia, 2008, 79, 179 CrossRef PubMed.
- H. Li and Z. Y. Wang, RSC Adv., 2015, 5, 30711 RSC.
- J. J. Yi, Z. Y. Wang, H. N. Bai, X. J. Yu, J. Jing and L. L. Zuo, Molecules, 2015, 20, 10450 CrossRef PubMed.
- P. Zou, X. Yang, W. W. Huang, H. T. Zhao, J. Wang, R. B. Xu, X. L. Hu, S. Y. Shen and D. Qin, Molecules, 2013, 18, 9933 CrossRef PubMed.
- L. J. Sun, Y. R. Guo, C. C. Fu, J. J. Li and Z. Li, Food Chem., 2013, 136, 1022 CrossRef PubMed.
- S. Fattahi, E. Zabihi, Z. Abedian, R. Pourbagher, A. A. Motevalizadeh, A. Mostafazadeh and H. A. Niaki, Int. J. Mol. Cell. Med., 2014, 3, 102 Search PubMed.
- I. Limem, H. Harizi, K. Ghedira and L. C. Ghedira, Immunopharmacol. Immunotoxicol., 2011, 33, 309 CrossRef CAS PubMed.
- Y. Yi, M. W. Zhang, S. T. Liao, R. F. Zhang, Y. Y. Deng, Z. C. Wei, X. J. Tang and Y. Zhang, Carbohydr. Polym., 2012, 87, 636 CrossRef CAS.
- S. H. Liu, J. H. Zhao, K. K. Deng, Y. Wu, J. W. Zhu, Q. H. Liu, H. H. Xu, H. F. Wu, X. Y. Li, J. W. Wang and Q. F. Guo, Spectrochim. Acta, Part A, 2015, 140, 202 CrossRef CAS PubMed.
- L. Vela, M. Contel, L. Palomera, G. Azaceta and I. Marzo, J. Inorg. Biochem., 2011, 105, 1306 CrossRef CAS PubMed.
- S. Y. Wang, Y. Liang and S. Zheng, J. Chromatogr. A, 2012, 1265, 46 CrossRef CAS PubMed.
- L. J. Sun, Y. R. Guo, C. C. Fu, J. J. Li and Z. Li, Food Chem., 2013, 136, 1022 CrossRef CAS PubMed.
- G. L. Nappo, A. Russo and V. Carbone, Food Chem., 2010, 123, 157 CrossRef.
- J. R. Araújo, P. Gonçalves and F. Martel, Nutr. Res., 2011, 31, 77 CrossRef PubMed.
- C. H. Huang, C. C. Huang, L. S. Hsu, S. H. Kao and C. J. Wang, J. Funct. Foods, 2015, 12, 80 CrossRef.
- J. F. Melissa, L. Joanne, F. L. Li, G. L. Li, H. Mikael and N. Patrick, PLoS One, 2011, 6, 1 Search PubMed.
- J. H. Chang, Y. Y. Shim, S. K. Cha and K. M. Chee, J. Appl. Microbiol., 2010, 109, 220 CrossRef CAS PubMed.
- C. Helma and M. Uhl, Mutat. Res., 2000, 466, 9 CAS.
- W. D. Ma, Y. P. Zou and P. Wang, Food Chem. Toxicol., 2014, 70, 1 CrossRef CAS PubMed.
- K. E. Hwang, C. Park, S. J. Kwon, Y. S. Kim, D. S. Park, M. K. Lee, B. R. Kim, S. H. Park, K. H. Yoon, E. T. Jeong and H. R. Kim, Int. J. Oncol., 2013, 43, 262 CAS.
- G. B. Park, Y. Choi, Y. S. Kim, H. K. Lee, D. Kim and D. Y. Hur, Int. J. Oncol., 2013, 43, 29 CAS.
- G. Kroemer, L. Galluzzi and C. Brenner, Physiol. Rev., 2007, 87, 99 CrossRef CAS PubMed.
- J. H. Lu, B. Quearry and H. Harada, FEBS Lett., 2006, 580, 3539 CrossRef CAS PubMed.
- S. Ghavami, M. Hashemi, S. R. Ande, B. Yeganeh, W. Xiao and M. Eshraghi, J. Med. Genet., 2009, 46, 497 CrossRef CAS PubMed.
- F. Hajiaghaalipour, M. S. Kanthimathi, J. Sanusi and J. Rajarajeswaran, Food Chem., 2015, 169, 401 CrossRef CAS PubMed.
- P. Darvin, Y. H. Joung, S. P. Nipin, D. Y. Kang, H. J. Byun, D. Y. Hwang, K. H. Cho, K. D. Park, H. K. Lee and Y. M. Yang, J. Funct. Foods, 2015, 15, 193 CrossRef CAS.
- H. Kim, J. Y. Moon, H. Kim, D. S. Lee, M. Cho and H. K. Choi, Food Chem., 2010, 121, 429 CrossRef CAS.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495 CrossRef CAS PubMed.
- H. H. Lin, K. C. Chan, J. Y. Sheu, S. W. Hsuan, C. J. Wang and J. H. Chen, Food Chem., 2012, 132, 880 CrossRef CAS.
- T. J. Sayers, Cancer Immunol. Immunother., 2011, 60, 1173 CrossRef CAS PubMed.
- Y. S. Ma, S. C. Hsu, S. W. Weng, C. C. Yu, J. S. Yang, K. C. Lai, J. P. Lin, J. G. Lin and J. G. Chung, Environ. Toxicol., 2015, 29, 1156 Search PubMed.
- D. Sareen, P. R. van Ginkel and J. C. Takach, Invest. Ophthalmol. Visual Sci., 2006, 47, 3708 Search PubMed.
- P. R. van Ginkel, S. R. Darjatmoko and D. Sareen, Invest. Ophthalmol. Visual Sci., 2008, 49, 1299 Search PubMed.
- J. H. Ahn, Y. I. Yang, K. T. Lee and H. Choi, J. Cancer Res. Clin. Oncol., 2015, 144, 255 CrossRef PubMed.
- Q. S. Xiang, Y. F. Ma, J. L. Dong and R. L. Shen, Int. J. Food Sci. Nutr., 2015, 66, 76 CrossRef CAS PubMed.
- R. H. Xu, H. Pelicano and Y. Zhou, Cancer Res., 2005, 65, 613 CrossRef CAS PubMed.
- A. C. Faber, F. J. Dufort, D. Blair, D. Wagner, M. F. Roberts and T. C. Chiles, Biochem. Pharmacol., 2006, 72, 1246 CrossRef PubMed.
- A. Kueck, A. W. Opipari and K. A. Griffith, Gynecol. Oncol., 2007, 107, 450 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.