
Stability of gum arabic-gold nanoparticles in physiological simulated pHs and their selective effect on cell lines†
Heloise Ribeiro de Barrosa,
Mateus Borba Cardosob,
Carolina Camargo de Oliveirac,
Célia Regina Cavichiolo Francoc,
Daniel de Lima Belanc,
Marcio Vidottia and
Izabel C. Riegel-Vidotti*a
aGrupo de Pesquisa em Macromoléculas e Interfaces, Departamento de Química, Universidade Federal do Paraná – UFPR, CxP 19032, CEP 81531-980, Curitiba, PR, Brazil. E-mail: izabel.riegel@ufpr.br
bBrazilian Synchrotron Light Laboratory, P.O. Box 6154, 13083-970, Campinas, Brazil. Fax: +55 19 3512-1004
cDepartamento de Biologia Celular, Universidade Federal do Paraná – UFPR, CxP 19081, CEP 81531-980, Curitiba, PR, Brazil
First published on 8th January 2016
Abstract
For the safe use of nanoparticles, especially in the biomedical field, their stability in different environments and the prevention of binding to the component organisms, which could lead to nanoparticle aggregation, is indispensable. Herein, we present a simple, efficient and biologically based method to obtain small gum arabic (GA)-stabilized gold nanoparticles (GA-AuNPs) with remarkable stability in physiological pHs. The in vitro stability tests in intestinal (pH 6.8) and gastric (pH 1.2) simulated pHs revealed that GA-AuNPs exhibit a surprisingly high stability even near the zero zeta potential. When subjected to GA-AuNPs, changes in the viability, proliferation and morphology were selectively induced in the B16-F10 melanoma cell line, whereas no alterations in the macrophage cell line, RAW 264.7, or in the fibroblast cell line, BALB/3T3, were observed at the same concentrations. Therefore, considering the remarkable stability and selective effect on cell lines, we show that GA-AuNPs exhibit properties that could provide a future alternative for melanoma treatment.
Introduction
Gold nanoparticles (AuNPs) have been widely explored because they exhibit singular properties depending on size, size distribution, shape, morphology and surface functionalization.1–4 Recent studies point to their potential biomedical applications, being promising in non-invasive strategies for cancer diagnosis and treatment.5,6 Additionally, AuNPs have been used to detect proteins or other molecules that can bind to gold surfaces.7,8 Among all of the properties, a good dispersion and the size stability both have a significant impact on the final therapeutic performance.9 Therefore, the development of reliable methodologies for the synthesis of AuNPs with a controllable shape, size and size distribution and that produce particles that are stable under various environments is very important from both a scientific and technological point of view.The most common employed strategy for obtaining AuNPs involves the nucleation of gold ions using a reducing agent to form nanoparticles.10 However, the presence of a stabilizing agent is necessary to provide more stable and relatively monodispersed AuNPs. Natural compounds have widely been employed either for the reduction of gold ions or for the protection of the formed gold nanoparticles in aqueous media.11,12 The advantages of using biomolecules include reduced toxicity and the ability to recognize specific biological targets.13
Polysaccharides have been widely employed for the preparation of hybrid gold and silver nanoparticles.14–16 Gum arabic (GA) is a highly branched, nontoxic and biocompatible polysaccharide that is naturally exudated from acacia trees trunks and barks. GA molecules contain galactose and arabinose as the main monosaccharide constituents as well as rhamnose and uronic acid in minor quantities.17 Additionally, glycoproteins and arabinogalactan-proteins (AGP) are present in a typical GA sample as the low and high molecular weight fractions, respectively. The existence of hydroxyl, carboxyl and amino acid groups makes GA molecules sensitive to the ionic strength and pH of the environment, providing a polyelectrolyte characteristic to GA. Therefore, the variety of functional groups in the GA chains and their complex microstructural molecular features are the key factors to the excellent performance of GA as a stabilizing agent for nanoparticles.18–21 Regarding the toxicity, Chanda et al.6,22 demonstrated that GA-AuNPs are biocompatible and do not induce mammalian cell toxicity, which suggests a good outlook for their future use in clinical applications.
Many techniques have been employed to determine the formation and the post-synthesis properties of gold nanoparticles. Among them, UV-Vis spectroscopy is a reliable technique that is used to monitor AuNP formation. In the UV-Vis spectrum, it is possible to observe an absorption band with a typical maximum wavelength (λmax) and peak intensity.3,23 The profile of the SPR (surface plasmon resonance) band is sensitive to the environmental pH that affects the surface charge distribution of the particles, resulting in a change in the oscillation of the surface electrons.24,25 Therefore, the SPR provides substantial information on the nanoparticle properties and has been successfully combined with imaging and scattering techniques to study a variety of nanoparticles in solution.3,26–29
In this work, we present the synthesis of AuNPs at room temperature and in the presence of GA. The formation of the metallic particles was shown using UV-Vis, whereas their shape, size and size distribution were determined using a combination of transmission electron microscopy and scattering techniques. Because we aim toward future biological applications, a detailed in vitro study was performed to understand the influence of the environmental pH on the stability of these GA-coated nanoparticles. Moreover, using in vitro assays, we analysed the induced effects promoted by the GA-AuNPs in melanoma (B16-F10), macrophage (RAW 264.7), and fibroblast (BALB/3T3) cell lines. The GA-AuNPs remained stable and were able to influence the melanoma viability and morphology with no harm to other cells. Our study confirms the safe use of gold nanoparticles in a variety of biological applications, and it sheds light on the possible selective antitumor activity of GA-AuNPs.
Experimental
Materials
GA was solubilized in water (18.2 MΩ cm, Millipore, USA), dialyzed for 48 h against distilled water through a dialysis membrane (12–14 kDa cut-off) and freeze-dried. Gold ions were obtained by dissolution of HAuCl4 (30% in dilute HCl) in a proper amount of water. The gastric (pH 1.2) and intestinal (pH 6.8) simulated pHs were obtained by preparing buffer solutions of KCl/HCl (ionic strength = 0.40 mol L−1) and KH2PO4/NaOH (ionic strength = 0.25 mol L−1), respectively. All reagents were purchased from Sigma-Aldrich unless otherwise stated.Preparation and characterization of the gold nanoparticles
The gum arabic-coated gold nanoparticles (GA-AuNPs) were obtained via reduction of AuCl4− in an aqueous medium using a one-pot method at 25 °C. Gold nanoparticles were successfully synthesized in aqueous medium, either in the presence of 0.1 wt% of GA (GA-AuNPs) or in the absence of GA (bare-AuNPs). The GA-AuNPs were obtained by reduction of AuCl4− in aqueous medium. For this, 5 mL of HAuCl4 1.75 × 10−4 mol L−1 were mixed, under magnetic stirring, with 5 mL of GA 0.1 wt% solution (previously left overnight at 4 °C to ensure complete hydration). Then, the reducing agent NaBH4 (10−3 mol L−1) was added at a rate of 200 μL/3 min. The total amount of NaBH4 solution was 2.4 mL. The AuNP formation was monitored using UV-Vis spectroscopy (Agilent model 8453) via the observation of the SPR band with a maximum at approximately λmax = 520–540 nm. A reducing agent was added until we observed two consecutive and coincident values for the λmax and absorbance intensity. The reaction was completed in less than 1 h. For comparison studies, the same procedure was conducted in the absence of GA or any other stabilizing agent (bare-AuNPs). We note that GA alone was not able to reduce Au(III) to Au(0) to form the AuNPs.Transmission electron microscopy (TEM) was performed using a JEOL 1200EX-II microscope at an acceleration voltage of 80 kV. A drop (∼10 μL) of the colloidal solution was deposited onto 400 mesh carbon-coated grids and air dried. The size and size distribution of the GA-AuNPs and bare-AuNPs were determined by observing 500 to 2000 objects from the same sample using ImageJ and OriginPro 8 software, respectively. Small-angle X-ray scattering (SAXS) measurements were performed on the D1B-SAXS1 beamline at the LNLS to determine the size, shape and polydispersity of the nanoparticles. The scattered X-ray beam with a wavelength (λ) of 1.55 Å was detected using a Pilatus 300k detector. The sample-to-detector distances were 942.7 mm and 3100.0 mm to cover a scattering vector, q (q = (4π/λ)sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ), ranging from 0.03 to 4 nm−1, respectively, where 2θ is the scattering angle. The measurements were performed at room temperature, and silver behenate was measured under the same conditions to calibrate the sample-to-detector distance, the detector tilt and the direct beam position. Transmission, dark current and mica sheet corrections were performed. The normalized scattering image of the samples was then subtracted from the normalized scattering image of pure water, and the isotropic result was radially averaged to obtain I(q) vs. q.
θ), ranging from 0.03 to 4 nm−1, respectively, where 2θ is the scattering angle. The measurements were performed at room temperature, and silver behenate was measured under the same conditions to calibrate the sample-to-detector distance, the detector tilt and the direct beam position. Transmission, dark current and mica sheet corrections were performed. The normalized scattering image of the samples was then subtracted from the normalized scattering image of pure water, and the isotropic result was radially averaged to obtain I(q) vs. q.
In vitro stability tests
GA-AuNPs and bare-AuNPs were studied when they were subjected to the simulated gastric and intestinal pHs. Three volume parts of the as-prepared bare-AuNPs or GA-AuNP dispersion were added to one part of the simulated fluid. After dilution, the measured pHs remained the same as that of the simulated fluids since their ionic strength are high. The samples were stored at 37 °C for 7 days. The SPR band profiles of the samples were obtained by recording the UV absorbance at 24 h intervals to observe the changes in the nanometric character of the particles. Samples corresponding to the first and seventh day were analysed using TEM. The surface charge of the particles was obtained using a Zetasizer Nano ZS instrument. The dispersions obtained just after synthesis were used as the control samples (pH ∼ 5) and studied according to the same protocols.Cell culture and treatment
B16-F10 murine melanoma (BCRJ 0046), BALB/3T3 (ATCC CCL-163), and RAW 264.7 (ATCC TIB-71) cells were maintained in complete medium, Dulbecco's Modified Eagle's Medium (DMEM-D5648), that was supplemented with 10% foetal bovine serum (Gibco), 1 U mL−1 penicillin, and 1 μg mL−1 streptomycin (Gibco) at 37 °C under 5% CO2. The cells were kept under 70% confluence for no more than 10 passages, and they were plated into 96-well plates (2 × 103 cells per well) and 24-well plates (1 × 104 cells per well) containing a sterile glass coverslip. After 6 hours, the cells were treated with the addition of 10, 20, 50, and 100 μL of a GA-AuNP sterile colloidal dispersion (filtered through a Millipore membrane with nominal pore size of 0.22 μm) for 48 hours before they were assayed. Approximately 0.04 wt% GA was dissolved in the same manner and was used as the control group.Viability and proliferation assays
Neutral red (NR) and thiazolyl blue tetrazolium bromide (MTT) viability assays were performed.30,31 After 48 h of treatment, the cells that were plated onto the 96-well plates were incubated with 100 μg mL−1 NR solution at 37 °C and under 5% CO2 for 2 h. The internalized dye was extracted with 100 μL of 50:1 ethanol:glacial acetic acid (Merck). Similarly, after treatment, a 0.45 mg mL−1 MTT solution was added, and the cells were incubated at 37 °C and under 5% CO2 for 3 h. The crystal products were dissolved in 100 μL dimethyl sulfoxide (Merck). Crystal violet (CV) was used to determine the cell proliferation.32 Afterwards, cells were fixed with a Cytofix solution (BD) for 30 minutes on ice and stained for 10 min with a 0.25 mg mL−1 crystal violet (EMS) solution. The stained cells were washed in distilled water and incubated in 33% acetic acid (Merck) under agitation for 30 minutes. The resulting absorbance was verified at 550 nm for NR and MTT and at 570 nm for CV using a microplate reader ELx800 (BioTek). All assays were performed in quadruplicate.
Morphological assays
Light, scanning, and confocal microscopy were used to determine whether the GA-AuNP colloidal dispersion had any effect on the B16-F10 cell morphology. Thus, after 48 h of treatment, the cells were processed as described below. A haematoxylin/eosin (H&E – EMS) stain was performed after 95% ethanol cell fixation. Dyes were added for 1 min and 30 s with an intermediate washing step (distilled water). Next, the coverslips were dehydrated in ethanol and xylol (Merck), and mounted with Permount (EMS) onto a glass slide. The B16-F10 cells were imaged using a 40× objective lens using a light microscope (LEICA DM IL).Scanning electron microscopy (SEM) was performed after 1 h fixation in Karnovsky fixative (2.5% glutaraldehyde, 4% paraformaldehyde, 1 mM CaCl2, in cacodylate buffer at pH 7.2 – EMS). The cells were post-fixed in 1% OsO4 (EMS), dehydrated in ethanol (Merck), CO2 critical point dried (CPD 010 030 – Balzers), covered in gold (SCD 030 – Balzers), and observed under a 5000× TESCAN VEGA 3 LMU.
The fluorescence labelling was achieved after cell fixation with 1% paraformaldehyde (EMS) in a phosphate buffer solution (PBS). Actin filaments were labelled with phalloidin-Alexa 488 (Invitrogen) for 20 minutes. The cells were washed with PBS, and the coverslips were mounted in Fluoromount containing DAPI (EMS). The slides were observed with 20× and 60× objectives using a confocal microscope, A1RSiMP (Nikon).
Statistical analyses
The biological tests were performed in quadruplicate. All data were submitted to the Shapiro–Wilk normality test, followed by the most appropriate test depending on the data normal or non-normal distribution. *p < 0.05.Results and discussion
Synthesis and characterization of the gold nanoparticles
Gold nanoparticles were successfully synthesized in an aqueous medium either in the presence of 0.1 wt% of GA (GA-AuNPs) or in the absence of the stabilizing agent (bare-AuNPs). The formation of gold nanoparticles was confirmed by the appearance of a band in the UV-Vis spectra at approximately 520–540 nm, which corresponds to the colour change from yellow to purple-red. The UV-Vis spectra of both of the final dispersions (pH ∼ 5) are presented in Fig. 1A. The absorption maximum (λmax) at 524 nm and 537 nm indicated that in both samples the particles are in the nanometric range. | ||
| Fig. 1 UV-Vis absorption spectra of bare-AuNPs (---) and GA-AuNPs 0.1 wt% (—) (A) and the corresponding zeta potential curves with Gaussian fits (B). | ||
Zeta potential (ζ) measurements of the bare-AuNP and GA-AuNPs were performed to correlate the GA presence and the overall surface charge of the nanoparticles (Fig. 1B). Bare and GA-AuNPs presented net negative surface charges ranging from −35 to −10 mV (ζmax = −22.3 mV) and from −50 to −10 mV (ζmax = −28.2 mV), respectively. For the bare-AuNPs, the source of the negative surface charge is more likely due to the adsorption of AuCl4−/AuCl2− ions. However, the origin of the negative charge for the GA-AuNPs is due to the GA-ionizable groups, such as carboxyl, from the acidic monosaccharides20 and amino acids from the protein fractions. Under the condition used in our experiment (pH = 5.5), the carboxyl groups are dissociated and the amino groups are protonated,33 resulting in a negatively charged surface of GA-AuNPs. Thus, from the point of view of the electrostatic interactions, both GA-AuNPs and bare-AuNPs are considered to be very stable.
TEM images were obtained to investigate the shape and size of the nanoparticles. As shown in Fig. 2A, the GA-AuNPs are mainly spherical. However, spherical bare-AuNPs are barely observable in Fig. 2B; however, a variety of other shapes are present. The average sizes were obtained through the best Gaussian fit of the size distribution curves, giving 5.4 ± 2.1 nm for the GA-AuNPs and 16.2 ± 6.1 nm for the bare-AuNPs. In the latter case, we measured the Feret diameter, which corresponds to the distance between two parallel lines restricting the largest dimension of the object. Therefore, as expected, the bare-AuNPs are larger, asymmetric and have a broader size distribution when compared to the GA-AuNPs.
 | ||
| Fig. 2 Representative TEM images and the corresponding particle size distribution profiles for the GA-AuNPs (A and C) and bare-AuNPs (B and D). The lines correspond to the best Gaussian fit. | ||
The concentration of the AuNPs dispersions, in particle per mL, was determined based on particle size and measured extinction coefficient obtained by UV-Vis measurements and applying the Lambert–Beer law. The concentration of GA-AuNPs is 9.90 × 1013 particles per mL and the concentration of bare-AuNP is 3.86 × 1011 particles per mL.
Because TEM measurements are highly dependent on the sample preparation and on the number of observed objects, a more representative and reliable technique was employed to characterize the particle sizes and morphology. Fig. 3 shows the SAXS patterns and their corresponding fits as well as the particle size distributions for the AuNPs synthesized in the presence and in the absence of GA. The scattering patterns presented in Fig. 3 were fit using a multi-structural level fit approach34 (solid lines), and two distinct regions were identified. The high-q region of the scattering curves (q > ∼0.4 nm−1) were attributed to the contribution of gold nanoparticles followed by the background.28 It was also fit using a polydisperse spherical form factor, shown as dashed lines in Fig. 3A and B.
 | ||
| Fig. 3 SAXS patterns of the AuNPs in the presence (A) and in the absence of GA (B) and their corresponding fits (solid lines). Scattering contributions from the gold nanoparticles are presented as dashed lines. The arrow in panel A highlights the scattering contribution due to the GA presence in the low-q region. The particle size distributions of the gold nanoparticles in the presence and in the absence of GA obtained from the dashed line SAXS fits are inserted. | ||
Regarding the low-q region (q < 0.2 nm−1), both samples presented a significant deviation from the spherical scattering contribution of the gold nanoparticles. In Fig. 3A, the deviation is attributed to the scattering due to the presence of GA. The same scattering profile is also observed in the same q range when a solution of GA was analysed (ESI, Fig. S1†). In both figures, the arrows indicate the common low-q region, which highlights the presence of the polymer in the scattering of the GA-AuNPs sample. However, the low-q deviation in Fig. 3B presents a different nature when compared to Fig. 3A. In this specific case, the deviation can be attributed to the presence of large aggregates, which are also observed in the TEM images. This aggregation behaviour was previously observed in other similar systems.24 No further information can be extracted from the SAXS due to the subtle deviation compared with the scattering of the spherical AuNPs. The log-normal size distributions were plotted between 0 and 30 nm to compare both samples. Comparatively, the size distribution obtained when GA was present (inset of Fig. 3A) is shifted to smaller values for the diameter when compared to the sample obtained in the absence of the polymer (inset of Fig. 3B). The average radius calculated from the fit of the GA-AuNPs is 1.82 nm (average diameter of approximately 4 nm), whereas for the bare-AuNPs, the average radius obtained from the fit is 6.2 nm (average diameter of approximately 12.5 nm). Therefore, the SAXS results are in good agreement with the TEM analysis because the dimensions obtained from both techniques are within the same size range.
In vitro stability studies
In vitro studies were conducted to evaluate the gold nanoparticle stability when subjected to the body-simulated pH at body temperature (37 °C). Additionally, these tests were designed to elucidate the origin of the superior stability of the GA-AuNPs. For comparative purposes, the stability behaviour of the bare-AuNPs was also investigated.Both samples were dispersed in simulated gastric and intestinal conditions at a pH of 1.2 and 6.8, respectively. The maintenance of the desired pH was verified daily. Fig. 4 displays the SPR band of the GA-AuNPs and bare-AuNPs in the different pHs for the study period. GA-AuNPs display a remarkable stability in all of the studied environments, as demonstrated by the retention of nearly the same λmax and FWMH values, as observed in Fig. 4A–C. Consequently, the stability of the dispersions was also verified by the maintenance of the characteristic red-purple colour. However, the bare-AuNPs show clear evidence of aggregation in the first 24 h at a pH of 5.5 (Fig. 4D) and 6.8 (Fig. 4E). This aggregation process is primarily observed by the shift in the λmax to higher wavelengths and by the increase in the SPR width. After the addition of the bare-AuNPs in the gastric fluid, the SPR instantaneously disappears (Fig. 4F) and the former red-purple solution changes to a colourless solution. These observations are evidence that the bare-AuNPs lose their nanometric properties, as previously expected, because the uncoated AuNPs are very unstable and prone to aggregate.
 | ||
| Fig. 4 UV-Vis absorption spectra of the GA-AuNPs (A–C) and bare-AuNPs (D–F) that were kept at 37 °C for seven days at the indicated pH. The arrows follow the time course. | ||
The TEM images that were obtained at the beginning (as-prepared) and at the end of the stability tests support the UV-Vis spectroscopy results. According to the Gaussian fit of the size distribution curve (ESI, Fig. S2†), the GA-AuNPs dispersed in the simulated intestinal fluid exhibited an average size of 4.8 ± 1.6 nm and 5.2 ± 1.9 nm at the beginning and at the end of the stability test, respectively. When the GA-AuNPs were dispersed in the simulated gastric fluid, the initial average diameter was 5.4 ± 1.6 nm and the final average diameter was 5.6 ± 1.6 nm. Then, the preservation of the particles sizes during the tests in both fluids was demonstrated.
Similarly, the same trend was observed when the GA-AuNPs were evaluated at a pH of 5.5. The TEM images also indicated that the spherical shape was preserved during the in vitro experiments (ESI, Fig. S3†). As verified by UV-Vis and TEM experiments, the GA-AuNPs remained very stable under the studied pHs at 37 °C during all seven days, which is an important result considering the biological applications due the strong dependence of the size and shape of the nanoparticles on their properties.
Zeta potential (ζ) measurements were performed to correlate the residual surface charge of the GA-AuNPs dispersed in the simulated fluids with the stability behaviour of the nanoparticles (Fig. 5). At pH 5.5, the ζ remained nearly constant (−23 mV) over the entire experimental period. At pH 6.8, a slight increase was observed in the third day (−19 mV), which remained constant thereafter. However, after the dispersion of the GA-AuNPs in the simulated gastric fluid (pH 1.2), the ζ shifted to approximately zero (−2 mV). The behaviour observed in Fig. 5 can be rationalized as follows. At any pH, the double layer thickness decreases with increasing ionic strength of the medium. As a consequence, the surface potential decreases, diminishing the stability of the colloidal particles. In our study, the solution at pH 5.5 has the lowest ionic strength, whereas the solution at pH 1.2 has the highest ionic strength, which is in agreement with the observed results.
 | ||
| Fig. 5 Zeta potential of the GA-AuNPs as a function of the exposure time at various pHs. | ||
Because the properties of the nanoparticles depend on their size, size distribution and morphology, it is of utmost importance to obtain nanoparticles that can maintain their properties in diverse environments. Additionally, it is of great interest to impart additional features to the nanometric particles via attachment to their surface molecules. Our study concludes that GA acts as an excellent stabilizing agent that maintains the gold nanoparticles properties in simulated gastric and intestinal pHs.
Regarding the formation of metallic nanoparticles, using the chemical reduction method, the first step corresponds to the nucleation period in which a new phase is formed followed by nuclei growth. Eventually, the environmental conditions promote interparticle interactions causing aggregation or breakage because the total potential energy for two interacting nanoparticles is the key factor that determines the final morphology of the aggregates. A commonly accepted mechanism for the bottom-up formation of stabilized gold colloidal dispersions involves the initial formation of a certain number of nuclei that adsorbs to the stabilizer molecular chain. The driving force for the nuclei adsorption is a decrease in the surface energy of gold atoms through favourable interactions with the stabilizer sites that alter the electron density distribution of the particles. After the nucleation period, an increase in the particle size occurs via the addition of new species to their surfaces. The classical nucleation and growth mechanism, which was first proposed by LaMer,35 says that a determined initial number of nuclei grow competitively via the near-equal division of available precursors, leading to particles with roughly the same size and narrow distribution.36 An improvement to LaMer's model considers the aggregation of small nanoparticles, which also participate in the growth process. In our study, because the reducing agent was added in aliquots during the course of the reaction, we assumed that nucleation and growth occurred simultaneously, providing particles with varied sizes of a broad distribution. This was only the case for the AuNPs in the absence of any stabilizing agent. However, when the formation of the gold nanoparticles was assisted by GA, despite the addition of the reducing agent in the aliquots, we found very small particles with a narrow size distribution. Consequently, the GA provides an excellent stabilizing environment either to the newly formed nuclei or to the formed particles, which results in the desired performance for controlling the NP synthesis. This is a very important result because monodisperse nanoparticles are sometimes desired because the dispersity controls the biodistribution37–39 and is also responsible for the nanoparticle biological functionalities.28
Based on the Derjaguin–Landau and Verwey–Overbeek (DLVO)40 theory, which considers that the colloidal stability is governed by the balance of the electrostatic repulsion with the van der Waals attraction, the mechanism of aggregation of a charged particle includes the increase of interparticle van der Waal's attractive forces due to the surface charge screening by the opposite sign species. Additionally, at any pH, an increase in the ionic strength of the colloidal gold solution reduces the electrical double layer thickness, favouring interparticle interactions and leading to aggregation. From an electrostatic interactions point of view, zeta potential values between −25 mV and +25 mV are typically related to very unstable systems.41–43 However, in our study, GA-AuNPs were not prone to aggregation, even at a nearly zero ζ, demonstrating that GA plays a role in the stabilization of the NPs that is not only related to the electrostatic mechanisms.
Thus, the stability of GA-AuNPs can be primarily attributed to the existence of steric forces that prevent nanoparticle aggregation independent of the pH because GA-AuNPs showed remarkable stability over time in an aqueous medium. For biological applications, steric stabilization is more advantageous than electrostatic stabilization because particle stability is maintained even in the presence of existing charges. Additionally, to minimize the interactions with biological membranes and improve the permeation, the particles are required to exhibit a neutral or slightly negative zeta potential.44
Selective changes induced by the GA-AuNPs in the melanoma cells
The potential application for GA-AuNPs was tested. First, we determined the cell viability using the NR and MTT assays after 48 h of treatment using different GA-AuNPs concentrations. NR is a hydrophilic dye that is internalized and then protonated in specific cellular compartments and becomes trapped inside viable cells.30 MTT can be easily reduced by mitochondrial enzymes when cells are viable, resulting in crystals that can be dissolved and assayed.31 B16-F10 cells treated with increasing GA-AuNPs concentrations exhibited decreased cell viability as a function of concentration (Fig. 6A and B). The IC50 was calculated for both assays, and it resulted in 142.7 and 80.2 for the NR and MTT assays, respectively. To determine if the gold nanoparticles affected the B16-F10 cells, we used an approximate average of the IC50 concentration (100 μL) by using only the 0.04 wt% GA (final concentration of GA in GA-AuNPs synthesis) for comparison. | ||
| Fig. 6 Selective changes induced by different volumes of GA-AuNPs in B16-F10 viability determinate through (A) IC50 in neutral red and MTT assays considering control group as 100% viable. The highest amount of GA-AuNPs (100 μL) was tested in (B) B16-F10, observing the decrease in cell viability **p < 0.005 and ***p < 0.0001, (C) BALB/3T3 and (D) RAW 264.7 cells observing no changes in neutral red and MTT viability assays. (E) Proliferation assay for B16-F10 cells induced by GA-AuNPs, which was decreased by 50 and 100 μL (***). T10, T20, T50 and T100 are referent to the cells treatments with 10, 20, 50 and 100 μL of GA-AuNPs. G100 is referent to the control with 100 μL of GA 0.04 wt%. | ||
The cell viability was decreased by the GA-AuNPs treatment (approximate 10%), which was promoted by the presence of the gold nanoparticle, with no interference from the stabilizing agent (GA).
Furthermore, to determine if the same behaviour would be observed in other cell lines, we assayed macrophage-like cells (RAW 264.7) and a non-tumorigenic fibroblast cell line (BALB/3T3). No differences were observed (Fig. 6C and D); thus, we could not determine IC50 for these cell lines (not shown). Therefore, among the cell lines tested, the GA-AuNPs decreased the cellular viability only for the B16-F10 tumorigenic cells.
Then, we determined whether B16-F10 proliferation was affected by the treatment. We found a decrease in cell proliferation after 50 and 100 μL of GA-AuNPs treatment (Fig. 6E) compared with GA alone. Thus, in addition to decreasing the B16-F10 cell viability, the GA-AuNPs are also able to reduce the cell proliferative behaviour of these cells.
The B16-F10 morphological properties were evaluated using different microscopy techniques (Fig. 7). Light microscopy showed that as the GA-AuNPs concentration increased, the cell number decreased (Fig. 7A), and the cytoplasmic extensions increased, which was also confirmed by SEM (Fig. 7B). Additionally, changes in the actin filament distribution were observed via confocal microscopy (Fig. 7C–E). Apparently, the cells were more spread out and adhered to the substrate, probably displaying areas of contact inhibition. This was clearly evident by the projection images (Fig. 7E) where the change in the cell nuclei distribution was observable. Control cells grew on the top of each other, whereas the treated cells did not. The text in Fig. 7E refers to the values obtained by the statistical analysis of the digitally processed images (width, height and depth of the plated cells).
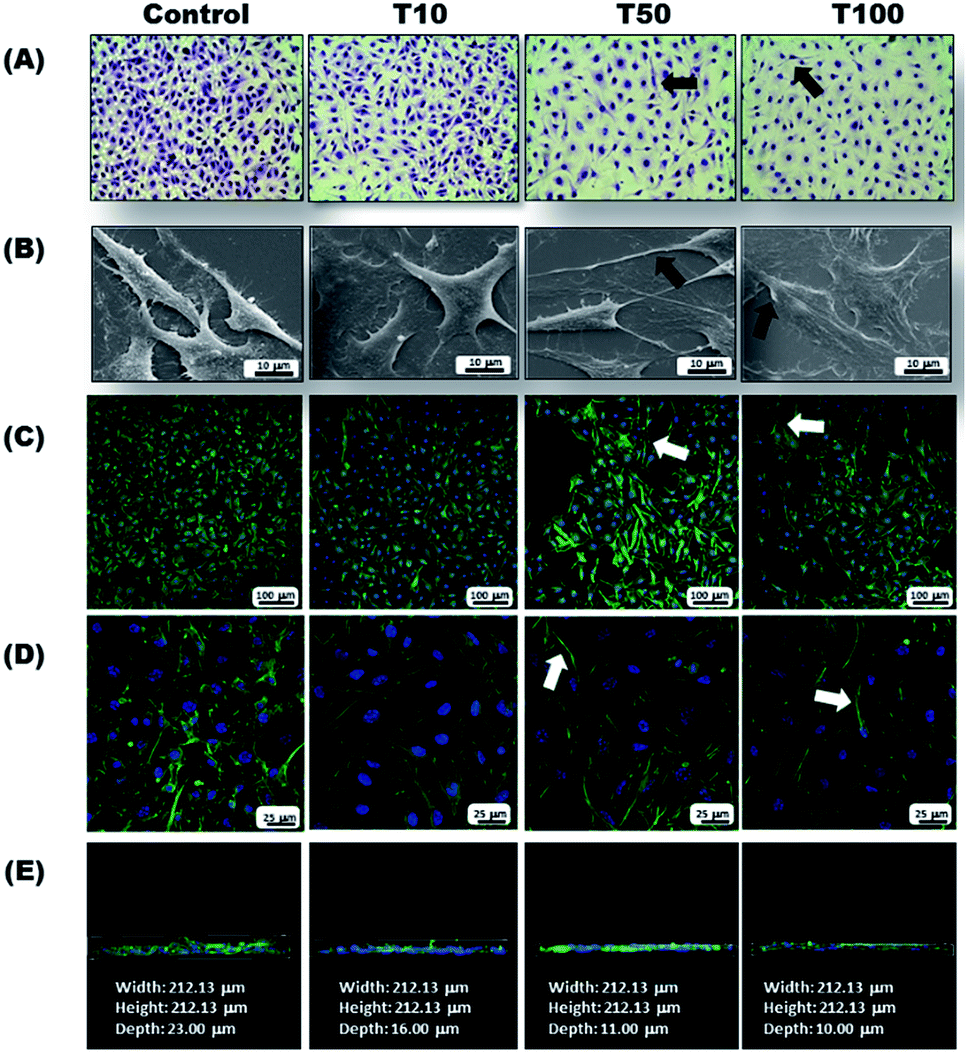 | ||
| Fig. 7 Images from morphological assays of B16-F10 exposed to 10, 50 and 100 μL of GA-AuNPs observed by different microscopy techniques. (A) Light microscopy (40×), (B) scanning electronic microscopy (7000×), scale bar = 10 μm, (C) and (D) confocal microscopy (20× and 60×, scale bars = 100 μm and 25 μm) and (E) projection of the (D) images onto the z axis. Note the reduction of cell number with treatment. Treated cells are more spread over and adhered on the substrate, also evidenced by cell nuclei distribution over the coverslip (depth values are 23, 16, 11 and 10 μm for the control, T10, T50 and T100, respectively). Arrows indicate morphological features discussed in the text. | ||
Melanoma is the most aggressive form of skin cancer, especially because of its metastatic ability. According to the World Health Organization, in 2012, approximately 55000 people died from melanoma, which was 119 people per 100000 habitants in Brazil.45 Although the U.S. Food and Drug Administration (FDA) has approved 6 new drugs for melanoma treatment in the past 4 years,46 research efforts are now focused on finding more efficient therapeutic approaches because the mortality and morbidity rates are still high.
Gold nanoparticles have demonstrated that can be used in disease diagnostic and therapeutic approaches.47–49 In particular, they exhibit remarkable features for cancer theranostics, varying from using them alone when applying laser therapy to many different types of molecular conjugations to their surfaces, increasing the therapy efficacy.50 In in vivo assays, gold nanoparticles have been shown to accumulate at the tumour sites due to the enhanced permeability and retention effect (EPR) and to change the angiogenesis and fenestration (gaps between endothelial cells) patterns, impairing tumour drug delivery51 but favouring nanoparticle selective retention and penetration into the tumour.52 Therefore, the tumour vasculature acts as a natural target with a high affinity for AuNPs.53,54 GA has been reported to functionalize radioactive AuNPs55 in the formation of gold nanocomposites in the presence of synchroton X-ray irradiated GA56 as well as to stabilize gold nanocrystals.6 These GA-AuNPs were implicated in cancer treatment and in vivo tumour tracking using X-rays. Although preliminary, the GA-AuNPs synthesized using our method exert a selective influence over melanoma cells, leading to a decrease in cell viability and proliferation as well as a change in the morphological properties that increases the cell adhesion. Here we described that stable gold nanoparticles coated with GA are able to promote selective changes in melanoma cells with no 2need to modify the nanoparticle surface or promote laser ablation or radiotherapy. Furthermore, our results demonstrated that the viability of other cells (macrophage cell RAW 264.7 and fibroblast BALB/3T3 cell lines) has not changed.
Promising new cancer treatments use molecules that target the immune system.57 GA has previously been described to have immunomodulatory properties by acting on dendritic cells.58 Polysaccharides extracted from similar sources were also implicated in macrophage stimulation59 with tumoricidal effects against Sarcoma-180.60 In our work, we did not assess the immunomodulatory properties; however, no cytotoxic effects were found when the macrophage cell line was treated with GA-AuNPs. This may suggest that GA-AuNPs are an excellent vehicle that may target tumorigenic cells without weakening the immunologic system, which could contribute to their ability to improve the stimulation and, even more, their efficacy against cancer cells.
Current general cancer treatment are aimed at proteins or signalling pathways that are shared with normal cells. Researchers are challenged to find less toxic approaches or selective ways to detect and control tumour cells. Efforts on therapies development that induce immune system, monoclonal antibodies targeting specific tumour proteins, or even cancer vaccines are being developed and some were recently approved for clinical use.57 In our work no viability changes were found when a macrophage cell line was treated with GA-AuNPs. Although we did not assess GA-AuNP immunomodulatory properties, GA alone was previously described to have immunomodulatory properties by acting on dendritic cells.58 Polysaccharides extracted from similar sources were also implicated in macrophage stimulation59 with tumoricidal effects against Sarcoma-180.60 This may suggest that GA-AuNPs are excellent vehicles that besides potentially targeting tumorigenic cells, could also stimulate the immune system contributing to a better outcome. Further in vitro and in vivo approaches should be considered to determine GA-AuNPs potential applications in cancer treatment.
Conclusions
Gold nanoparticles that were stabilized by gum arabic were obtained using a simple and efficient methodology under mild synthesis conditions. The procedure described here is not dependent on further purification or fractionation steps prior to the application of the nanoparticles. The obtained gum GA-AuNPs are of a small size and narrow size distribution because GA acts as an effective stabilizer that controls the formation of the AuNPs during their synthesis. The in vitro stability study confirmed that the colloidal stabilization was promoted by GA in the intestinal (pH = 6.8) and gastric (pH = 1.2) simulated pHs at 37 °C. The stability of the GA-AuNPs is attributed primarily to the existence of steric forces that prevent the nanoparticle aggregation, regardless of the environmental pH. Additionally, when tested in the presence of the GA-NPs, selective changes in the viability, proliferation and morphology were observed in melanoma cells. Other cells (macrophage cell RAW 264.7 and fibroblast BALB/3T3 cell lines) were tested comparatively and presented no alteration. Although further investigations are necessary, our study contributes to the possible use of the studied nanoparticles in cancer immunotherapy.Acknowledgements
The authors gratefully acknowledge the Brazilian funding agency, CNPq (grants 477467/2010-5 and 470691/2010-7), Rede Nanoglicobiotec/CNPq (475641/10-08) and Fapesp (project numbers 2011/21954-7 and 2014/22322-2) for financial support. The LNLS is acknowledged for SAXS measurements (project number 10783) and the D1B-SAXS1 beamline staff for the assistance during experiments. We also sincerely thank Prof. Elisa S. Orth and Prof. Marcela Mohallem Oliveira for fruitful discussions. Gabriela Ematsu and Jessica Fernanda Affonso de Oliveira are also acknowledged for their guidance during the zeta-potential measurements. We would also like to thank the Instituto Carlos Chagas for kindly providing BALB/3T3 and Raw 264.7 cells. Finally, the authors are very grateful to the Electron Microscopy Center of UFPR (CME-UFPR) for the SEM images.References
- R. Sardar, A. L. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS PubMed.
- C. J. Orendorff, A. Gole, T. K. Sau and C. J. Murphy, Anal. Chem., 2005, 77, 3261–3266 CrossRef CAS PubMed.
- M. Vidotti, R. F. Carvalhal, R. K. Mendes, D. C. M. Ferreira and L. T. Kubota, J. Braz. Chem. Soc., 2011, 22, 3–20 CrossRef CAS.
- L. Treuel, M. Malissek, J. S. Gebauer and R. Zellner, ChemPhysChem, 2010, 11, 3093–3099 CrossRef CAS PubMed.
- N. Chanda, P. Kan, D. W. Lisa, R. Shukla, A. Zambre, T. L. Camarck, H. Engelbrecht, J. R. Lever, K. Katti, G. M. Fent, S. W. Casteel, J. Smith, W. H. Miller, S. Jurisson, E. Boote, D. Robertson, C. Cutler, M. Dobrovolskaia, R. Kannan and K. V. Katti, Nanomedicine, 2010, 6, 201–209 CrossRef CAS PubMed.
- N. Chanda, A. Upendran, E. J. Boote, A. Zambre, S. Axiak, K. Selting, K. V. Katti, W. M. Leevy, Z. Afrasiabi, J. Vimal, J. Singh, J. C. Lattimer and R. J. Kannan, Biomed. Nanotechnol., 2014, 10, 383–392 CrossRef CAS.
- H. Guan, J. Yu and D. Chi, Food Control, 2013, 32, 35–41 CrossRef CAS.
- R. Hermann, P. Walther and M. Muller, Histochem. Cell Biol., 1996, 106, 31–39 CrossRef CAS PubMed.
- S. Du, K. Kendall, P. Toloueinia, Y. Mehrabadi, G. Gupta and J. Newton, J. Nanopart. Res., 2012, 14, 758 CrossRef.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef PubMed.
- S. Si, M. Raula, T. K. Paira and T. K. Mandal, ChemPhysChem, 2008, 11, 1578–1584 CrossRef PubMed.
- Y. C. Wang and S. J. Gunasekaran, J. Nanopart. Res., 2012, 14, 1200 CrossRef.
- X. Liu, H. Huang, G. Liu, W. Zhou, Y. Chen, Q. Jin and J. Ji, Nanoscale, 2013, 5, 3982–3991 RSC.
- Y. Park, Y. N. Hong, A. Weyers, Y. S. Kim and R. J. Linhardt, IET Nanobiotechnol., 2011, 5, 69–78 CrossRef CAS PubMed.
- Z. Shervani and Y. Yamamoto, Carbohydr. Res., 2011, 346, 651–658 CrossRef CAS PubMed.
- L. F. de Oliveira, J. O. de Gonçalves, K. A. de Gonçalves, J. Kobarg and M. B. Cardoso, J. Biomed. Nanotechnol., 2013, 9, 1817–1826 CrossRef PubMed.
- P. A. Williams and G. O. Phillips, Handbook of Hydrocolloids, Woodhead Publishing Limited, Cambridge, 2000 Search PubMed.
- D. K. Devi, S. V. Pratap, R. Haritha, K. S. Sivudu, P. Radhika and B. Sreedhar, J. Appl. Polym. Sci., 2010, 121, 1765–1773 CrossRef.
- R. C. Quintanilha, E. S. Orth, A. Grein-Iankovski, I. C. Riegel-Vidotti and M. Vidotti, J. Colloid Interface Sci., 2014, 434, 18–27 CrossRef CAS PubMed.
- P. A. Cornelsen, R. C. Quintanilha, M. Vidotti, P. A. J. Gorin, F. F. Simas-Tosin and I. C. Riegel-Vidotti, Carbohydr. Polym., 2015, 119, 35–43 CrossRef CAS PubMed.
- I. Rocha, E. Lucht, I. C. Riegel-Vidotti, M. Vidotti and E. S. Orth, J. Phys. Chem. C, 2014, 118, 25756–25764 CAS.
- S. Vijayakumar and S. Ganesan, J. Nanomater., 2012, 2012, 1687–4110 Search PubMed.
- J. Doak, R. K. Gupta, K. Manivannan, K. Ghosh and P. K. Kahol, Phys. E, 2010, 42, 1605–1609 CrossRef CAS.
- J. F. A. Oliveira and M. B. Cardoso, Langmuir, 2014, 30, 4879–4886 CrossRef PubMed.
- K. Nakamura, T. Kawabata and Y. Mori, Powder Technol., 2003, 131, 120–128 CrossRef CAS.
- E. Boisseler and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- P. Mulvaney, Langmuir, 1996, 12, 788–800 CrossRef CAS.
- V. Dal Lago, L. F. de Oliveira, K. L. Gonçalves, J. Kobarg and M. B. Cardoso, J. Mater. Chem., 2011, 21, 12267–12273 RSC.
- H. R. Luckarift, S. Balasubramanian, S. Paliwal, G. R. Johnson and A. L. Simonian, Colloids Surf., B, 2007, 58, 28–33 CrossRef CAS PubMed.
- E. Borenfreund and J. A. Puerner, Toxicol. Lett., 1985, 24, 119–124 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- B. Bonnekoh, A. Wevers, F. Jugert, H. Merk and G. Mahrle, Arch. Dermatol. Res., 1989, 281, 487–490 CrossRef CAS PubMed.
- B. C. da Silva, M. de Oliveira, J. G. L. Ferreira, M. R. Sierakowski, F. F. Simas-Tosin, E. S. Orth and I. C. Riegel-Vidotti, Food Hydrocolloids, 2015, 46, 201–207 CrossRef CAS.
- G. Beaucage, J. Appl. Crystallogr., 1995, 28, 717–728 CrossRef CAS.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- S. P. Shields, V. N. Richards and W. E. Buhro, Chem. Mater., 2010, 22, 3212–3225 CrossRef CAS.
- B. Pelaz, S. Jaber, D. J. Aberasturi, V. Wulf, T. Aida, J. M. Fuente, J. Feldmann, H. E. Gaub, L. Josephson, C. R. Kagan, N. A. Kotov, L. M. Liz-Marzan, H. Mattoussi, P. Mulvaney, C. B. Murray, A. L. Rogach, P. S. Weiss, I. Willner and W. J. Parak, ACS Nano, 2012, 6, 8468–8483 CrossRef CAS PubMed.
- J. H. Kim, J. H. Kim, K. W. Kim, M. H. Kim and Y. S. Yu, Nanotechnology, 2009, 20, 50510 Search PubMed.
- L. C. Estrada and E. Gratton, ChemPhysChem, 2012, 13, 1087–1092 CrossRef CAS PubMed.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 1992 Search PubMed.
- T. C. Prathna, N. Chandrasekaran, A. M. Raichur and A. Mukherjee, Colloids Surf., A, 2011, 377, 212–216 CrossRef CAS.
- T. Kim, K. Lee, M. Gong and S. W. Joo, Langmuir, 2005, 21, 9524–9528 CrossRef CAS PubMed.
- T. Kim, C. H. Lee, S. W. Joo and K. J. Lee, J. Colloid Interface Sci., 2008, 318, 238–243 CrossRef CAS PubMed.
- C. M. Dawidczyk, C. Kim, J. H. Park, L. M. Russell, K. H. Lee, M. G. Pomper and P. C. Searson, J. Controlled Release, 2014, 187, 133–144 CrossRef CAS PubMed.
- B. W. Stewart and C. P. Wild, World cancer report 2014, International Agency for Research on Cancer, Lyon, 2014 Search PubMed.
- J. A. Lo and D. E. Fisher, Science, 2014, 346, 945–949 CrossRef CAS PubMed.
- R. R. Letfullin, C. B. Iversen and T. F. George, Nanomedicine, 2011, 7, 137–145 CrossRef CAS PubMed.
- L. Bobyk, M. Edouard, P. Deman, M. Vautrin, K. Pernet-Gallay, J. Delaroche, J. F. Adam, F. Estève, J. L. Ravanat and H. Elleaume, Nanomedicine, 2013, 9, 1089–1097 CrossRef CAS PubMed.
- J. P. M. Almeida, E. R. Figueroa and R. A. Drezek, Nanomedicine, 2014, 10, 503–514 CrossRef CAS PubMed.
- R. M. Cabral and P. V. Baptista, Expert Rev. Mol. Diagn., 2014, 14, 1041–1052 CrossRef CAS PubMed.
- M. C. F. Simões, J. J. S. Sousa and A. A. C. C. Pais, Cancer Lett., 2015, 357, 8–42 CrossRef PubMed.
- S. Nazir, T. Hussain, A. Ayub, U. Rashid and A. J. MacRobert, Nanomedicine, 2014, 10, 19–34 CrossRef CAS PubMed.
- K. V. Katti, R. Kannan, K. Katti, V. Kattumori, R. Pandrapraganda, V. Rahing, C. Cutler, E. J. Boote, S. W. Casteel, C. J. Smith, J. D. Robertson and S. S. Jurrison, Czech J. Phys., 2006, 56, 23–56 CrossRef.
- R. Bhattacharya and P. Mukherjee, Adv. Drug Delivery Rev., 2008, 60, 1289–1306 CrossRef CAS PubMed.
- R. Kannan, A. Zambre, N. Chanda, R. Kulkarni, R. Shukla, K. Katti, A. Upendran, C. Cutler, E. Boote and V. K. Katti, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 42–51 CrossRef CAS PubMed.
- X. Liu, H. Huang, G. Liu, W. Zhou, Y. Chen, Q. Jin and J. Ji, Nanoscale, 2013, 5, 3982–3991 RSC.
- A. Karlitepe, O. Ozalp and C. B. Avci, Tumor Biol., 2015, 36, 4075–4078 CrossRef CAS PubMed.
- N. T. Xuan, E. Shumilina, O. Nasir, D. Bobbala, F. Götz and F. Lang, Cell. Physiol. Biochem., 2010, 25, 641–648 CrossRef PubMed.
- M. P. Moretão, D. L. Buchi, P. A. J. Gorin, M. Iacomini and M. B. M. Oliveira, Immunol. Lett., 2003, 89, 175–185 CrossRef.
- M. P. Moretão, A. R. Zampronio, P. A. J. Gorin, M. Iacomini and M. B. M. Oliveira, Immunol. Lett., 2004, 93, 189–197 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra24858b |
| This journal is © The Royal Society of Chemistry 2016 |