
Microwave-assisted synthesis of 3-sulfenylindoles by sulfonyl hydrazides using organic ionic base-Brønsted acid†
Rajjakfur Rahaman,
Namita Devi,
Kuladip Sarma and
Pranjit Barman*
Department of Chemistry, National Institute of Technology Silchar, Silchar 788010, India. E-mail: barmanpranjit@yahoo.co.in; Fax: +91 3842 224797; Tel: +91 9435374128
First published on 21st January 2016
Abstract
A novel, efficient, and high-yielding method was developed for the synthesis of 3-sulfenylindoles via a DBU-based ionic liquid promoted sulfenylation of indoles using sulfonyl hydrazides as a thiol surrogate. The environmentally friendly procedure, easy operation and mild reaction conditions enable the tolerance of a wide scope of functionalities as well as high reaction efficiency. The synthetic procedure is suitable for both N-protected or unprotected indoles.
Introduction
Sulfenylated indole moieties represent a class of very important organosulfur heterocyclic compounds as they are present in many pharmaceutically and biologically important molecules.1 Among the various indole derivatives, 3-sulfenylindoles have significant interest due to their greater therapeutic value in the treatment of several diseases (Chart 1), for example, HIV (1),2 cancer (2),3 vascular (3),4 respiratory disorders (4),5 heart disease6 and allergies.7 They are also used as potent inhibitors of both tubulin polymerization and of cancer cells.8 These attractive biological profiles are the basic cause of long standing interest in the development of efficient methods for the synthesis of 3-sulfenylindoles. In the last few decades, a number of significant methods have been developed for the synthesis of 3-sulfenylindoles. A variety of sulfenylating reagents have been discovered as reaction partners during the synthetic efforts. The synthesis of 3-sulfenylindoles included the direct sulfenylation of the indole ring by sulfenyl halides,9 disulfides,10 thiols,11 quinine mono-O,S-acetals,12 arylsulfonyl chlorides,13 N-thioimides,14 and sulfonium salts.15 Nevertheless, many of these thiolating agents are air and moisture sensitive, toxic, expensive, or possess unpleasant odors. Moreover, previously reported methodologies for the sulfenylation of indoles have some practical limitations, such as long reaction time, harsh reaction conditions, or excess sulfenylating agents, high temperature, or yield byproducts unfriendly to the environment. | ||
| Chart 1 Some biologically active 3-arylthioindoles. | ||
Recently, sulfonyl hydrazide16 has emerged as a new and efficient sulfenylating agent. These are readily accessible, odorless, and easy to handle and exist as stable solids. They have been satisfactorily used as reductants,17 sulfonyl sources18 through the cleavage of their sulfur–nitrogen bonds, and aryl sources19 through the cleavage of their carbon–sulfur bonds. They are satisfactorily used in microwave assisted synthesis as a sulfenylating agent.20
In the current green chemistry scenario, microwave-assisted organic synthesis (MAOS) has attained the status of a new and fascinating discipline.21 Microwave synthesis has been reduce reaction times dramatically, increase product yields and purities by reducing unwanted side reactions compared to conventional heating methods.22 The domain was further strengthened by ionic liquids (ILs) owing to their-c eco-safe properties.23 1,8-Diazabicycloundec-7-ene (DBU) is widely used as an organic base,24a–c and DBU-based ionic liquids, such as [DBU][HOAc], are particularly useful as a non-nucleophilic task-specific organic ionic base.20b,24d
Since the energy of the microwave photon is very low, the enhancement of reaction rate is the combination of thermal and non-thermal effects of microwave radiation.25 The mechanism of microwave effect must be consistent with the physics of relaxation process in solution and resonant process in gaseous phase. Moreover, the reactions that take place in solution are primarily affected by the change of dipole moments of the solute, solvent, and the catalyst. The physics of microwave interaction with molecules in solution is followed by the change in polarization from dipole orientation, which is demonstrated by Debye dielectric relaxation theory.26
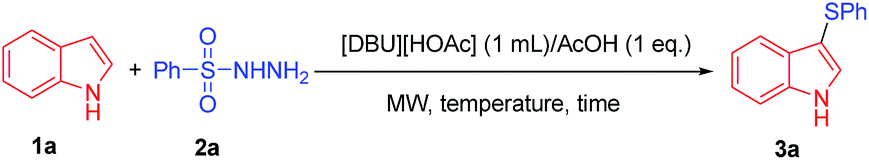
According to the present synthetic requirements, application of microwave (MW) synthetic methodology using ionic liquids is particularly welcome.27,20b Hence it is imperative to exploit the combination of IL and MW for organic transformations with careful assessment of a pair of distinct organo systems in different permutations. Herein, we report a green protocol for the 3-sulfenylation of indoles with sulfonyl hydrazides as thiol surrogate under microwave irradiation (Scheme 1).
 | ||
| Scheme 1 DBU-based ionic liquid 3-sulfenylation of indoles. | ||
Results and discussion
At the outset of this study, we employed indole 1a and benzenesulfonyl hydrazide 2a as the model substrate in the presence of [DBU][HOAc] ionic base and AcOH (Table 1). The reaction was carried out in various duration of reaction time as 3, 5, 7 and 10 min and out of this at 10 min afforded the desired product in almost quantitative yield (Table 1, entry 4).
|
||||
|---|---|---|---|---|
| Entry | MW (W) | T (°C) | Time (min) | Yieldb (%) |
| a Reaction conditions: indole 1a (0.5 mmol), benzenesulfonyl hydrazide 2a (0.6 mmol), [DBU][HOAc] (1 mL), AcOH (1 equiv.), 10 min.b Isolated yield.c Conventional heating (open vessel reflux condition).d Reaction performed without microwave irradiation at room temperature (closed vessel). | ||||
| 1 | 80 | 100 | 3 | 40 |
| 2 | 80 | 100 | 5 | 65 |
| 3 | 80 | 100 | 7 | 75 |
| 4 | 80 | 100 | 10 | 92 |
| 5 | 80 | 110 | 10 | 80 |
| 6 | 80 | 90 | 10 | 70 |
| 7 | 100 | 100 | 10 | 92 |
| 8 | 60 | 100 | 10 | 60 |
| 9 | — | 100 | 18 h | 75c |
| 10 | — | r.t. | 40 h | 50d |
In the next step, we tested the effect of temperature and influence of the microwave irradiation power in various levels. However, at 100 °C and 80 W provided the desired product 3a in 92% yield. Increasing the power from 80 to 100 W did not affect the reaction since the product 3a was obtained in the same yield (Table 1, entry 7). However, on decreasing the power to 60 W the yield decreased considerably (Table 1, entry 8). A temperature higher or lower than 100 °C was deleterious to the reaction (Table 1, entries 5 and 6). Moreover, we examined the reaction under conventional heating in oil bath and at room temperature giving the desired product 3a only 75% and 50% but required a very long reaction time (Table 1, entry 9 and 10).
After that, DBU and different ionic liquids were screened to improve the yield of the product. We examined number of ionic liquids such as Bmim[BF4], Bmim[Br], Bmim[OH] and [DBU][HOAc] (Table 2, entries 1 to 5), out of these non-nucleophilic ionic base, [DBU][HOAc] afforded the desired product with high yield. Other DBU based ionic liquids like [DBU][n-Pr], [DBU][n-Bu] and [DBU][TFA], were evaluated but not effective like [DBU][HOAc] (Table 2, entries 6–8).
|
||||
|---|---|---|---|---|
| Entry | 1a/2a | Organic ionic base | Additive (equiv.) | Yieldb (%) |
| a Reaction conditions: indole 1a, benzenesulfonyl hydrazide 2a, [DBU][HOAc] (1 mL), AcOH, 10 min.b Isolated yield based on 1a. | ||||
| 1 | 0.5/0.5 | DBU | 15 | |
| 2 | 0.5/0.5 | Bmim[BF4] | 10 | |
| 3 | 0.5/0.5 | Bmim[Br] | 0 | |
| 4 | 0.5/0.5 | Bmim[OH] | 20 | |
| 5 | 0.5/0.5 | [DBU][HOAc] | 45 | |
| 6 | 0.5/0.5 | [DBU][n-Pr] | 35 | |
| 7 | 0.5/0.5 | [DBU][n-Bu] | 25 | |
| 8 | 0.5/0.5 | [DBU][TFA] | 0 | |
| 9 | 0.5/0.6 | [DBU][HOAc] | 55 | |
| 10 | 0.5/0.6 | [DBU][HOAc] | AcOH (1.5) | 92 |
| 11 | 0.5/0.6 | [DBU][HOAc] | AcOH (1) | 70 |
| 12 | 0.5/0.6 | [DBU][HOAc] | AcOH (2) | 85 |
| 13 | 0.5/0.6 | [DBU][HOAc] | DABCO (1.5) | 50 |
| 14 | 0.5/0.6 | [DBU][HOAc] | L-Proline (1.5) | 55 |

The effect of additive was also monitored to look into the effect of cooperative operation. We examined different additives namely AcOH, DABCO and L-proline; it was observed that AcOH provided the desired product in high yield when used in stoichiometric amount. The best molar ratio of indole/sulfonyl hydrazide was found to be 0.5/0.6 (Table 2). Therefore, the standard reaction conditions for the synthesis of 3-sulfenylindoles were obtained: indole 1a (0.5 equiv.), benzenesulfonyl hydrazide 2a (0.6 equiv.), AcOH (1.5 equiv.) in [DBU][HOAc] (1 mL) at 100 °C and 80 W for 10 min.
With our optimized reaction conditions in hand (Table 2, entry 4), scope and limitations of the proposed method were investigated as shown in Table 3. First, we examined the substrate scope of sulfonyl hydrazides towards indole (Table 3, 3a–3j). A variety of sulfonyl hydrazides with electron donating and electron withdrawing groups were smoothly reacted with the indole to form their corresponding sulfenylated product in moderate to excellent yields (Table 3). The arylsulfonyl hydrazides with electron donating groups like –Me, –OMe on the phenyl ring produced the desired products with higher yields than those with electron withdrawing groups (–Cl, –Br, and –NO2). Next, we investigate the substrate scope of variously substituted indoles (Table 3, 3k–3r). Here we also got the same trend, indoles with electron donating groups like –Me, Ph were gave the desired products in better yields than those with electron withdrawing groups like –Cl, –Br, –NO2. It was found that C-2 substituted electron donating indoles delivered relatively higher yield compared with their 5-analogue. 2-Position of the indole ring becomes the active reaction site when 3-position is occupied by alkyl groups like –Me (Table 3, 4a). N-Substituted indoles also gave the corresponding desired products with high yields without any difficulties (Table 3, 3l). The recyclability of the ionic liquid was also checked. After completion of the reaction, the aqueous layer containing [DBU][HOAc] was dried and washed with diethyl ether. The recovered ionic liquid was used up to five times without any significant loss of activity.
|
|---|
| a Reaction conditions: indole 1 (0.5 mmol), sulfonyl hydrazide 2 (0.6 mmol), [DBU][HOAc] (1 mL), AcOH (1.5 mmol), MW (80 W), 10 min, 100 °C. |
 |

To demonstrate the synthetic utility of the new method, gram scale reaction was carried out under the optimized conditions (Scheme 2). Thereby, the reaction between 1H-indole 1a (5 mmol) and benzenesulfonyl hydrazides 2a (6 mmol) in the presence of [DBU][HOAc] (20 mL) and AcOH (10 mmol) afforded the desired product 3a in 85% yield after 1 hour of reaction time. Therefore, this method could be used to synthesize the precursors of many important biologically active molecules.
 | ||
| Scheme 2 Scale-up reaction between 1a and 2a. | ||
In order to gain insight of the mechanism, a control experiment was carried out under the optimized conditions with p-nitrophenylsulfonyl hydrazide, in absence of indole, which led to the formation of corresponding disulfide.20b The resulting disulfide separately made to react with indole 1a under the optimized procedure; it gave rise to the desired product 3e with a diminished yield.
On the basis of existing liturature,16a,20b control experiments and above experimental results, we proposed a plausible mechanism for 3-sulfenylation of indoles is outlined in Scheme 3. Initially, sulfonyl hydrazide (2) is reduced to form D in the presence of [DBU][HOAc] and AcOH. In the next step D is attacked by the indole to form F, it is formed through the path a, or path b. F release an H+ ion and gives the desired product 3.
 | ||
| Scheme 3 Proposed mechanism for the 3-sulfenylation of indoles. | ||
Conclusions
In summary, we have developed a practical microwave-assisted synthetic approach for the rapid and efficient sulfenylation of indoles with sulfonyl hydrazides through the cleavage of sulfur oxygen and sulfur nitrogen bonds. A range of aryl- and alkylsulfonyl hydrazides smoothly react with indoles to give 3-sulfenylindoles in moderate to excellent yields. Due to high yield, broad substrates scope, short reaction time and use of a recyclable ionic liquid, the process promises to be a greener alternative. The enhancement of reaction rate under microwave condition might be the combination of thermal and non-thermal effects to obtain the desired product. Microwave radiation is a very polarizing field and may stabilize polar transition states and intermediates, which accelerate the reaction rate by decreasing activation energy. However, more research is needed for understanding the effect of microwave rate enhancement. Further studies on the application of indole and sulfonyl hydrazides are ongoing in our laboratory.Experimental
General methods
1H and 13C NMR spectra were recorded in CDCl3 and DMSO-d6 at 600 MHz, 400 MHz, 150 MHz, 125 MHz and 100 MHz respectively. Chemical shifts (δ) are reported parts per million (ppm) and are referenced to tetramethylsilane (TMS) as internal standard. NMR multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, m = multiplet, br = broad signal. Chemical shifts for DMSO-d6 were reported at 3.3 and 2.45 ppm respectively (δ). All the synthesis processes were performed on a programmed microwave synthesis reactor (START SYNTH, Milestone), which is equipped with inner symmetrical glass tubes. The tubes were located on a rotated plate, which could make all the reactions in the same condition. The temperature was monitored by an inner IR detector. All the reaction parameters were programmed with optimized increased time, target temperature, standing time and temperature. TLC was done on silica gel coated glass slide (Merck silica gel G for TLC). For column chromatography silica gel 60–120 mesh (SRL, India) was used. Elemental analyses were performed on a Flash 2000 Thermo Scientific instrument at NIT Silchar. The yields are based on isolated compounds after purification. Melting points were recorded on an electro thermal digital melting point apparatus and were uncorrected.Reagents
All the chemicals/reagents including indoles, sulfonyl chlorides were purchased from Alfa Aesar, Sigma-Aldrich and E. Merck; and were used without further purification. New reagent bottles (Aldrich) of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) [≥99.0%], BF3·OEt2 [≥46.5% BF3 basis], 1,4-diazabicyclo[2.2.2]octane (DABCO) [≥99.0%], L-proline [≥98.0%], 1-butyl-3-methylimidazolium hexafluorophosphate (Bmim[PF6]) [≥98.5%], 1-butyl-3-methylimidazolium bromide (Bmim[Br]) [≥97.0%], and acetic acid (≥99.99%, based on metal analysis) were used. Sulfonyl hydrazides, Bmim[OH], [DBU][HOAc], [DBU][n-Pr], [DBU][n-Bu] and [DBU][TFA] were prepared according to the literature procedures.28–30General procedure for synthesis of 3-sulfenylindoles
To a mixture of indole 1 (0.5 mmol), sulfonyl hydrazide 2 (0.6 mmol), [DBU][HOAc] (1 mL) and AcOH (1.5 mmol) were taken in glass tubes, sealed and placed in the cavity of microwave apparatus. Then set parameters are as follows: microwave irradiation power 80 W, increasing time 5 min, target temperature 100 °C, standing time 10 min, standing temperature 100 °C. A maximum irradiation power of 80 W and 100 °C were applied for 10 min. After the temperature reached 100 °C, the instrument was automatically adjusted to maintain a constant temperature. After 10 min (monitored through TLC), the reaction mixture was cooled to room temperature, then diluted with distilled water and the organic layer was extracted with ethyl acetate (3 × 10 mL). The combined organic phase was washed with brine (3 × 10 mL). Dried over anhydrous Na2SO4 and the solvent was removed under vacuum. The crude product was purified by column chromatography on silica gel, eluting with petroleum ether/ethyl acetate (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), affording the desired product 3.
:1), affording the desired product 3.
Acknowledgements
The authors wish to thank All India Council of Technical Education (AICTE), New Delhi [8023/BOR/RID/RPS (NER)-68/2010–11] for financial support for this work. MHRD, Govt. of India is acknowledged for the doctorate fellowship (MHRD GATE fellowship) received by R. F. R. and N. D. Authors are also thankful to CSIR-NEIST Jorhat and IIT Guwahati for spectral analysis.Notes and references
- (a) R. L. Sundberg, Indoles, Academic, London, 1996 Search PubMed; (b) A. Casapullo, G. Bifulco, I. Bruno and R. Riccio, J. Nat. Prod., 2000, 63, 447 CrossRef CAS PubMed; (c) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed; (d) A. R. Katritzky and A. F. Pozharskii, Handbook of Heterocyclic Chemistry, Pergamon, Oxford, 2000 Search PubMed; (e) B. Bao, Q. Sun, X. Yao, J. Hong, C. O. Lee, C. J. Sim, K. S. Im and J. H. Jung, J. Nat. Prod., 2005, 68, 711 CrossRef CAS PubMed; (f) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS PubMed; (g) M. C. van Zandt, M. L. Jones, D. E. Gunn, L. S. Geraci, J. H. Jones, D. R. Sawicki, J. Sredy, J. L. Jacot, A. T. Dicioccio, T. Petrova, A. Mischler and A. D. Podjarny, J. Med. Chem., 2005, 48, 3141 CrossRef CAS PubMed; (h) T. R. Garbe, M. Kobayashi, N. Shimizu, N. Takesue, M. Ozawa and H. Yukawa, J. Nat. Prod., 2000, 63, 596 CrossRef CAS PubMed.
- R. Ragno, A. Coluccia, G. La Regina, G. de Martino, F. Piscitelli, A. Lavecchia, E. Novellino, A. Bergamini, C. Ciaprini, A. Sinistro, G. Maga, E. Crespan, M. Artico and R. Silvestri, J. Med. Chem., 2006, 49, 3172 CrossRef CAS PubMed.
- G. La Regina, M. C. Edler, A. Brancale, S. Kandil, A. Coluccia, F. Piscitelli, E. Hamel, G. de Martino, R. Matesanz, J. F. Díaz, A. I. Scovassi, E. Prosperi, A. Lavecchia, E. Novellino, M. Artico and R. Silvestri, J. Med. Chem., 2007, 50, 2865 CrossRef CAS PubMed.
- N. Zhou, W. Zeller, M. Krohn, H. Anderson, J. Zhang, E. Onua, A. S. Kiselyov, J. Ramirez, G. Halldorsdottir, T. Andresson, M. E. Gurney and J. Singh, Bioorg. Med. Chem. Lett., 2009, 19, 123 CrossRef CAS PubMed.
- D. Ainge, M. Butters, E. Merifield, R. Ramakrishnan, R. N. Rayapati, P. R. Sharma and C. Thomson, PCT Int. Appl. WO 2011004182 A1, January 13, 2011.
- C. D. Funk, Nat. Rev. Drug Discovery, 2005, 4, 664 CrossRef CAS PubMed.
- P. C. Unangst, D. T. Connor, S. R. Stabler, R. J. Weikert, M. E. Carethers, J. A. Kennedy, D. O. Thueson, J. C. Chestnut, R. L. Adolphson and M. C. Conroy, J. Med. Chem., 1989, 32, 1360 CrossRef CAS PubMed.
- (a) G. de Martino, G. La Regina, A. Coluccia, M. C. Edler, M. C. Barbera, A. Brancale, E. Wilcox, E. Hamel, M. Artico and R. Silvestri, J. Med. Chem., 2004, 47, 6120 CrossRef CAS PubMed; (b) G. de Martino, M. C. Edler, G. La Regina, A. Coluccia, M. C. Barbera, D. Barrow, R. I. Nicholson, G. Chiosis, A. Brancale, E. Hamel, M. Artico and R. Silvestri, J. Med. Chem., 2006, 49, 947 CrossRef CAS PubMed; (c) G. La Regina, M. C. Edler, A. Brancale, S. Kandil, A. Coluccia, F. Piscitelli, E. Hamel, G. de Martino, R. Matesanz, J. F. Díaz, A. I. Scovassi, P. Ennio, A. Lavecchia, E. Novellino, M. Artico and R. Silvestri, J. Med. Chem., 2007, 50, 2865 CrossRef CAS PubMed.
- (a) P. Hamel, J. Org. Chem., 2002, 67, 2854 CrossRef CAS PubMed; (b) M. Raban and L.-J. Chern, J. Org. Chem., 1980, 45, 1688 CrossRef CAS.
- (a) L.-H. Zou, J. Reball, J. Mottweiler and C. Bolm, Chem. Commun., 2012, 48, 11307 RSC; (b) G. La Regina, V. Gatti, V. Famiglini, F. Piscitelli and R. Silvestri, ACS Comb. Sci., 2012, 14, 258 CrossRef CAS PubMed; (c) W. Ge and Y. Wei, Synthesis, 2012, 934 CAS; (d) W. Ge and Y. Wei, Green Chem., 2012, 14, 2066 RSC; (e) Z. Li, J. Hong and X. Zhou, Tetrahedron, 2011, 67, 3690 CrossRef CAS; (f) X.-L. Fang, R.-Y. Tang, P. Zhong and J.-H. Li, Synthesis, 2009, 4183 CAS; (g) P. Sang, Z. Chen, J. Zoua and Y. Zhang, Green Chem., 2013, 15, 2096 RSC; (h) Ch. D. Prasad, S. Kumar, M. Sattar, A. Adhikary and S. Kumar, Org. Biomol. Chem., 2013, 11, 8096 Search PubMed; (i) R. Rahaman, N. Devi and P. Barman, Tetrahedron Lett., 2015, 56, 4224 CrossRef CAS.
- (a) J. S. Yadav, B. V. S. Reddy, Y. J. Reddy and K. Praneeth, Synthesis, 2009, 1520 CrossRef CAS; (b) J. S. Yadav, B. V. S. Reddy and Y. J. Reddy, Tetrahedron Lett., 2007, 48, 7034 CrossRef CAS; (c) Y. Maeda, M. Koyabu, T. Nishimura and S. Uemura, J. Org. Chem., 2004, 69, 7688 CrossRef CAS PubMed; (d) K. M. Schlosser, A. P. Krasutsky, H. W. Hamilton, J. E. Reed and K. Sexton, Org. Lett., 2004, 6, 819 CrossRef CAS PubMed; (e) J. A. Campbell, C. A. Broka, L. Gong, K. A. M. Walker and J.-H. Wang, Tetrahedron Lett., 2004, 45, 4073 CrossRef CAS; (f) Y. Liu, Y. Zhang, C. Hu, J.-P. Wana and C. Wen, RSC Adv., 2014, 4, 35528 RSC; (g) G. Wu, J. Wu, J. Wu and L. Wu, Synth. Commun., 2008, 38, 1036 CrossRef CAS; (h) Y. Liu, Y. Zhang, C. Hu, J.-P. Wan and C. Wen, RSC Adv., 2014, 4, 35528 RSC.
- (a) M. Matsugi, K. Murata, H. Nambu and Y. Kita, Tetrahedron Lett., 2001, 42, 1077 CrossRef CAS; (b) M. Matsugi, K. Murata, K. Gotanda, H. Nambu, G. Anilkumar, K. Matsumoto and Y. Kita, J. Org. Chem., 2001, 66, 2434 CrossRef CAS PubMed.
- Q. Wu, D. Zhao, X. Qin, J. Lan and J. You, Chem. Commun., 2011, 47, 9188 RSC.
- (a) E. Marcantoni, R. Cipolletti, L. Marsili, S. Menichetti, R. Properzi and C. Viglianisi, Eur. J. Org. Chem., 2013, 132 CrossRef CAS; (b) C. C. Silveira, S. R. Mendes, L. Wolf and G. M. Martins, Tetrahedron Lett., 2010, 51, 2014 CrossRef CAS; (c) M. Tudge, M. Tamiya, C. Savarin and G. R. Humphrey, Org. Lett., 2006, 8, 565 CrossRef CAS PubMed.
- S. Jain, K. Shukla, A. Mukhopadhyay, S. N. Suryawanshi and D. S. Bhakuni, Synth. Commun., 1990, 20, 1315 CrossRef CAS.
- (a) F.-L. Yang and S.-K. Tian, Angew. Chem., Int. Ed., 2013, 52, 4929 CrossRef CAS PubMed; (b) A. K. Bagdi, S. Mitra, M. Ghosh and A. Hajra, Org. Biomol. Chem., 2015, 13, 3314 RSC; (c) X. Zhao, L. Zhang, X. Lu, T. Li and K. Lu, J. Org. Chem., 2015, 80, 2918 CrossRef CAS PubMed.
- (a) D. J. Pasto and R. T. Taylor, Org. React., 1991, 40, 91 CAS; (b) D. J. Pasto, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 8, p. 471 Search PubMed.
- (a) X. Li, X. Xu and C. Zhou, Chem. Commun., 2012, 48, 12240 RSC; (b) T. Taniguchi, A. Idota and H. Ishibashi, Org. Biomol. Chem., 2011, 9, 3151 RSC; (c) I. V. Ukrainets, A. A. Tkach, V. V. Kravtsova and S. V. Shishkina, Chem. Heterocycl. Compd., 2008, 44, 677 CrossRef CAS; (d) S. Kamijo, M. Al-Masum and Y. Yamamoto, Tetrahedron Lett., 1998, 39, 691 CrossRef CAS; (e) R. Ballini, E. Marcantoni and M. Petrin, Tetrahedron, 1989, 45, 6791 CrossRef CAS; (f) A. Deavin and C. W. Rees, J. Chem. Soc., 1961, 4970 RSC.
- (a) F.-L. Yang, X.-T. Ma and S.-K. Tian, Chem.–Eur. J., 2012, 18, 1582 CrossRef CAS PubMed; (b) B. Liu, J. Li, F. Song and J. You, Chem.–Eur. J., 2012, 18, 10830 CrossRef CAS PubMed; (c) X. Yu, X. Li and B. Wan, Org. Biomol. Chem., 2012, 10, 7479 RSC; (d) O. Y. Yuen, C. M. So, W. T. Wong and F. Y. Kwong, Synlett, 2012, 2714 CAS.
- (a) N. Singh, R. Singh, D. S. Raghuvanshi and K. N. Singh, Org. Lett., 2013, 15, 5874 CrossRef CAS PubMed; (b) R. Singh, D. S. Raghuvanshi and K. N. Singh, Org. Lett., 2013, 15, 4202 CrossRef CAS PubMed; (c) R. Singh, B. K. Allam, N. Singh, K. Kumari, S. K. Singh and K. N. Singh, Org. Lett., 2015, 17, 2656 CrossRef CAS PubMed.
- (a) B. Gutmann, A. M. Schwan, B. Reichart, C. Gspan, F. Hofer and C. O. Kappe, Angew. Chem., Int. Ed., 2011, 50, 7636 CrossRef CAS PubMed; (b) J. D. Moseley and C. O. Kappe, Green Chem., 2011, 13, 794 RSC.
- Microwave Assisted Organic Synthesis, ed. J. P. Tierney and P. Lidstroem, Blackwell, Oxford, 2005 Search PubMed.
- (a) T. Torimoto, T. Tsuda, K.-I. Okazaki and S. Kuwabata, Adv. Mater., 2010, 22, 1196 CrossRef CAS PubMed; (b) J. W. Lee, J. Y. Shin, Y. S. Chun, H. B. Jang, C. E. Song and S. G. Lee, Acc. Chem. Res., 2010, 43, 985 CrossRef CAS PubMed; (c) J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508 CrossRef CAS PubMed; (d) K. Dong and S. Zhang, Chem.–Eur. J., 2012, 18, 2748 CrossRef CAS PubMed.
- (a) C. Larrivee-Aboussafy, B. P. Jones, K. E. Price, M. A. Hardink, R. W. McLaughlin, B. M. Lillie, J. M. Hawkins and R. Vaidyanathan, Org. Lett., 2010, 12, 324 CrossRef CAS PubMed; (b) S. L. Peterson, S. M. Stucka and C. J. Dinsmore, Org. Lett., 2010, 12, 1340 CrossRef CAS PubMed; (c) N. de Rycke, F. Couty and O. R. P. David, Chem.–Eur. J., 2011, 17, 12852 CrossRef CAS PubMed; (d) A. G. Ying, L. Liu, G. F. Wu, G. Chen, X. Z. Chen and W. D. Ye, Tetrahedron Lett., 2009, 50, 1653 CrossRef CAS.
- (a) A. de la Hoz, A. Diaz-Ortiz and A. Moreno, Chem. Soc. Rev., 2005, 34, 164 RSC; (b) C. O. Kappe, B. Pieber and D. Dallinger, Angew. Chem., Int. Ed., 2013, 52, 1088 CrossRef CAS PubMed.
- G. B. Dudley, R. Richertb and A. E. Stiegman, Chem. Sci., 2015, 6, 2144 RSC.
- (a) V. Polshettiwar and R. S. Varma, Acc. Chem. Res., 2008, 41, 629 CrossRef CAS PubMed; (b) B. D. Narhe, M.-H. Tsai and C.-M. Sun, ACS Comb. Sci., 2014, 16, 421 CrossRef CAS PubMed.
- A. G. Myers, B. Zheng and M. Movassaghi, J. Org. Chem., 1997, 62, 7505 CrossRef.
- B. C. Ranu and S. Banerjee, Org. Lett., 2005, 7, 3049 CrossRef CAS PubMed.
- A.-G. Ying, L.-M. Wang, H.-X. Deng, J.-H. Chen, X.-Z. Chen and W.-D. Ye, ARKIVOC, 2009, xi, 288 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of synthesized compounds. See DOI: 10.1039/c5ra24851e |
| This journal is © The Royal Society of Chemistry 2016 |