
Phase transfer preparation of ultrasmall MnS nanocrystals with a high performance MRI contrast agent†
Jing Meng,
Yizhe Zhao,
Zhongfeng Li*,
Ligang Wang and
Yang Tian*
Department of Chemistry, Beijing Key Laboratory for Optical Materials and Photonic Devices, Capital Normal University, Beijing 100048, PR China. E-mail: lizf@cnu.edu.cn; tianyang@cnu.edu.cn
First published on 5th January 2016
Abstract
In this paper, a phase transfer method is reported which was used to prepare ultrasmall manganese(II) sulfide (MnS) nanocrystals (approximately 5 nm) in which prefabricated MnS aggregations are transferred from cyclohexane into an aqueous solution of sodium citrate. The MnS nanocrystals capped with citrate were extremely stable in the aqueous solution. Furthermore, the smart MnS nanocrystals showed super good behavior as MRI contrast agents. The relaxivity of the nanocrystals at a low magnetic field was 3.4 mM−1 s−1, which was higher than most reported values for manganese oxide. The MnS nanocrystal agent could produce highly efficient T1-weighted magnetic resonance images in vivo at a low dose, which is only 1/8–1/16 that of the commercial contrast agent gadolinium–diethylenetriamine penta-acetic acid. The MnS nanocrystals clearly enhanced the magnetic resonance imaging (MRI) contrast for use with the liver and kidney in vivo. The metabolic process of MnS nanocrystals was also studied. The in vivo results demonstrate that the simply prepared and smart MnS nanocrystals had a high performance and were promising candidates for MRI T1 contrast agents.
Introduction
Magnetic resonance imaging (MRI), which offers high spatial resolution and deep tissue penetration, is widely applied for diagnosis and post-therapy evaluation.1–7 To enhance the contrast in MRI images, paramagnetic compounds with a large number of unpaired electrons are desirable as contrast agents because they can shorten the longitudinal relaxation time (T1) and the transverse relaxation time (T2).8–10 T1-weighted MRI sequences are capable of accumulating in organs causing their images to become bright; therefore, T1 contrast agents are typically more preferred than T2 agents in the clinical setting.11–13 At present, gadolinium (Gd)-based contrast agents have been studied widely in clinical settings as T1 contrast agents. However, the extremely high toxicity of Gd-based agents can induce nephrogenic systemic fibrosis in patients with impaired kidney function and lead to brain abnormalities in healthy patients.14–18 Recent investigations have also demonstrated that magnetic iron oxide nanocrystals smaller than 5 nm are potentially useful as T1 contrast agents because their paramagnetism becomes dominant for small particles, but the T1 effects were relatively weak.19–23 Thus, a safer and more effective T1 contrast agent is still needed for MRI improvements in clinical science.The transition metal ion, manganese (Mn2+) with five unpaired electrons can also produce efficient positive contrast enhancement and offers an alternative MRI contrast agent.24 Mn has lower toxicity because Mn is a trace element and presents in an ionic form in the human body.13,25,26 For example, manganese(II) chloride (MnCl2) salt is even used as one of the examples of a Mn-based contrast agent approved by the FDA.27 From the point of view of safety and practicality, it is better to develop Mn-based nanomaterials for the ideal contrast enhancement with a low dose. At present, although Mn-based nanocrystals have successfully been used as T1 contrast agents, they are mainly composed of manganese oxide (MnO) nanocrystals.28–35 Furthermore, the higher relaxivity and stability of Mn-based contrast agents are still desirable for better imaging quality and reduced risk of misdiagnosis. Manganese sulfides (MnS) are a class of materials with potential for use in magnetic and optical applications.36 MnS exists in three different crystalline forms as a function of temperature: α-MnS (cubic), β-MnS (defect spinel), and γ-MnS (layered structure), in which α-MnS is the most stable.37–39 Specifically, MnS-suspended colloids (0.1–10 μm particles) are one of the earliest studied inorganic T1 agents.40 However, no reports were available on using their nanocrystals as contrast agents.
The efficiency of T1 contrast agents depends mainly on the longitudinal relaxivity r1, which is the rate of proton relaxation of the longitudinal process.41 Paramagnetic ions interact with the surrounding water protons and shorten the T1 relaxation time to enhance its relaxivity.11 The particle size is a major parameter for relaxivity enhancement. With a decreased particle size, the surface area of nanocrystals has increased dramatically. The rationale of using ultrasmall nanocrystals as T1 contrast agents lies in the strong surface effect on their magnetic properties, leading to a strong paramagnetic signal.28,42 In addition, the large surface areas also increase the water diffusion around and interaction with the particle surface, making ultrasmall nanoparticles good candidates for T1 MRI contrast agents.30,43–45 For example, Das et al. synthesized ultrasmall uniform sodium gadolinium fluoride (NaGdF4) nanocrystals with tunable sizes, and r1 relaxivity increased dramatically with decreasing particle size, from a value of 3.0 mM−1 s−1 for the 8.0 nm particles to 7.2 mM−1 s−1 for the smallest particles (i.e., 2.5 nm).41 Na et al. synthesized different sized MnO nanocrystals, and the r1 increased with decreasing particle size, from a value of 0.12 mM−1 s−1 for nanoparticles of 25 nm to 0.37 mM−1 s−1 for particles of 7 nm.13 Furthermore, it has recently been demonstrated that the nanoparticles with a size less than 10 nm are easily taken up and excreted, and show longer blood circulation times in comparison with larger ones.46
To date, although some hydrolytic synthetic methods and thermal decomposition in polar solvents can produce water-soluble nanocrystals, the preparation of high quality nanocrystals (e.g., highly monodisperse, uniform sized, controllable, and highly crystalline) relies on the organic solvent system with the assistance of hydrophobic surface capping agents.47 The prepared hydrophobic nanocrystals in the non-polar system limited their use in the biological environment.48,49 Therefore, the subsequent surface decoration by an amphiphilic polymer, surfactant or inorganic silica is obligatory before MRI contrast application.28,50–53 However, the surface decoration is typically accompanied by colloidal aggregation potential because of a low binding affinity.54,55 In particular, the decoration reduces relaxivity because of the long-chain capping agent which is used to prevent the water protons from interacting with the magnetic nanocrystal surface.
In, the first example of using MnS nanocrystals as a potential T1 contrast agent via a phase transfer method is reported: MnS nanocrystals of approximately 5 nm were produced by transferring the prefabricated MnS aggregations from cyclohexane into a aqueous sodium citrate solution. The MnS nanocrystals that were capped with citrate were extremely stable in the aqueous solution. Apart from their ultrasmall size and super hydrophilicity, the smart MnS nanocrystals behaved well in the contrast agent for MRI: they expressed a much higher relaxivity at the low magnetic field than most values reported for MnO. The highly efficient T1-weighted MR images in vivo were obtained using the MnS nanocrystal agent at a low dose, which is only 1/8–1/16 of the commercial contrast agent Gd–diethylenetriamine penta-acetic acid (Gd–DTPA). The metabolic process of the MnS nanocrystals in the kidney and liver was also studied. The in vivo results demonstrate that the easily prepared and smart MnS nanocrystals had a high performance and were promising candidates for MRI T1 contrast agents.
Experimental section
Chemicals
1-Octanol and cyclohexane were of analytical grade and obtained from Tianjin Guangfu Co. Ltd. Manganese chloride (MnCl2, 99%), thioacetamide (TAA, 99%), n-octylamine (99% mass fraction), and oleic acid were supplied by J&K Scientific Ltd. Acetone, ethanol (99%) and trisodium citrate (Na3Cit) were analytical grade and purchased from Beijing Chemical Company. Bovine serum albumin (BSA) was provided by Genview. All the chemicals were used as received without further purification. Water used in all experiments was purified using a water purification system (Pure Technology Co., Ltd) with a resistance higher than 18 MΩ cm.Preparation of MnS nanoaggregations
In a typical procedure to prepare MnS nanoaggregations, MnCl2 (0.0252 g, 0.2 mmol) and TAA (0.0452 g, 0.6 mmol) were dissolved in 3 mL of 1-octanol, 3 mL of n-octylamine, 3 mL of acetone and 1 mL of oleic acid with agitation for 10 min at room temperature. After formation of an homogeneous solution, it was transferred into a Teflon lined stainless steel autoclave with a capacity of 20 mL and bubbled with nitrogen for about 5 min at room temperature. The Teflon autoclave was sealed in a stainless autoclave and maintained at 150 °C for 1 h in a preheated oven. After the reaction, the Teflon autoclave was taken out and cooled down to the room temperature. The precipitate at the bottom was separated by centrifugation with a speed of 5500 rpm. The precipitate obtained was dispersed in 2 mL of cyclohexane at room temperature before further use.Phase transfer preparation of ultrasmall MnS nanocrystals
After 0.125 g sodium citrate was dissolved in 10 mL deionized water, 0.7 mL MnS–cyclohexane solution was added to it and the mixture was stirred vigorously at 85 °C for 15 min to remove the cyclohexane. The aqueous solution of MnS nanocrystals were obtained through the simple phase transfer process and then stored in an ampoule bottle under ambient conditions before further use in in vitro and in vivo magnetic resonance (MR) images.Relaxivity measurement
To evaluate the effect of the MnS nanocrystals as an MRI contrast agent, longitudinal relaxation times T1 of samples and the T1-weighted images with a series of Mn concentrations (0–0.6 mM Mn) were measured on a MRI Analyzing and Imaging system Micro MR20-025H-I 0.47 Tesla MRI scanner (Shanghai Niumag Corporation) at 32 °C. The r1 water proton relaxivities of the sample were then estimated from the slopes of 1/T1 versus the molar Mn concentration. T1-weighted images were acquired using a fast spin-echo sequence with the following parameters: FOV read = 100 mm, FOV phase = 100 mm, repetition time (TR) = 295 ms, echo time (TE) = 18.2 ms, slice width = 4.0 mm, slice gap = 1.0 mm, NS = 8, matrix size = 192 × 256. The sample was diluted in deionized water and BSA before longitudinal relaxation times T1 and the T1-weighted images were measured.Cell culture
Human hepatocellular carcinoma cells (Hep G2 cells) were grown in Dulbecco's modified Eagle's medium (DMEM, HyClone HIGH Glucose SH30022.01) culture growth medium, supplemented with 10% fetal bovine serum (FBS; v/v). The cells were incubated at 37 °C with 5% carbon dioxide (CO2) in a humidified environment.In vitro cytotoxicity assay
The cytotoxicity of the MnS nanocrystals was evaluated by using a cell viability assay with 4′,6-diamidino-2-phenylindole (DAPI) as stain. Hep G2 cells were seeded into a 12-well plate with 10% FBS at 37 °C and 5% CO2, and the following experiment was conducted until the 80% confluence point was reached. Subsequently, various concentrations of the MnS nanocrystals (0 μg mL−1, 10 μg mL−1, 30 μg mL−1, 50 μg mL−1, 70 μg mL−1) were injected onto a 12-well plate that was pre-seeded with the Hep G2 cells. The cells were then incubated at 37 °C for 12 h under the same conditions. A control group was treated with culture medium. All the tests were carried out in quintuplicate. To test for the cell viability, DAPI was added to each well and the 12-well plate was further incubated for 1 h before the fluorescent morphology images were taken using the Olympus IX71 fluorescence microscope. Apoptotic cell death was determined morphologically using the fluorescent nuclear dye (DAPI). The fluorescent morphology images showed the number of apoptotic cells with nuclear condensation and fragmentation. The control group was identified as 100% and the values of treated groups were normalized to the control group. The experimental values (treated) were expressed as a percentage of the control results.In vivo T1 MRI measurement
MRI in vivo was performed with a 0.5 T MesoMR-60 MRI system (Shanghai Niumag Corporation). Nude mice with body weight of ∼20 g were purchased by the Spectro Experimental Center and their use was approved by the Institute's animals' care and use committee. The mouse was anesthetized and maintained at normal body temperature. Subsequently, the MnS nanocrystals were injected via the tail vein into the mouse at a dose of 12.5 μmol kg−1 of weighed mouse. Longitudinal cross-sectional scan images were taken before and after the MnS nanocrystal administration for MRI analysis. T1-weighted fast spin-echo sequence MR images were acquired under the following parameters: H = 0.5 Tesla, temperature (T) = 32 °C, FOV read = 100 mm, FOV phase = 100 mm, TR = 300 ms, TE = 13.5 ms, slice width = 3.0 mm, slice gap = 0.5 mm, NS = 8, the matrix size = 192 × 256.Statistical method
The signal intensities were measured in the defined region of the interest (ROI) using ImageJ software. After normalization of the signal intensities, the intensity enhancement (IE) of ROI at time pointed t was calculated using eqn (1):| IE (%) = 100 × (ROI(t) − ROI(0))/ROI(0) | (1) |
It was hypothesised that the uptake and the excretion of the agent follow a simplified first-order reaction, i.e., the IE in the T1-weighted images may be expressed using eqn (2):
| IEt = IEmax × [1 − e−t/t1/2] | (2) |
| IEobsd = IEuptake − IEexcretion = IEmax × [−et/t1/2,uptake + e−t/t1/2,excretion] | (3) |
Characterization
Powder X-ray diffraction (XRD) patterns were collected using a Rigaku D/Max-2200 PC diffractometer with CuKα radiation (λ = 1.5418 Å) and a graphite monochromator from 20 to 70° with a scanning rate of 5.0° per min. Unit cell dimensions were determined using the JADE 5 program for XRD pattern processing, identification, and quantification. The size and morphology of the obtained cubic MnS were characterized using transmission electron microscopy (TEM, Jeol JEM-100CXII) equipped with a selected area electron diffractometer (SAED). The structure and lattice fringes of the nanocrystals were observed by using a high resolution TEM (HRTEM, FEI Tecnai G2 F30) equipped with a scanning transmission electron microscopy. The X-ray photoelectron spectroscopy (XPS) analyses were performed to characterize the components and surface of the surface of the cubic MnS. The XPS spectra were recorded on a spectrometer (PerkinElmer PHI-5300 ESCA) with its energy analyzer working in the pass energy mode at 35.75 eV, and the AlKa line was used as the excitation source. After subtraction of the X-ray satellites and inelastic background (Shirley-type), peak deconvolutiom was carried out. The XPS information obtained was based on the C–C standard peak (284.6 eV). The chemical composition of the as-prepared MnS was determined using energy dispersive X-ray spectroscopy (EDX, Oxford Instruments). The infrared spectrum [Nicolet 5DX Fourier-transform infrared (FTIR) instrument, potassium bromide (KBr) pellet technique] was carried out to characterize the surface of the cubic MnS. The Mn2+ concentration in the aqueous solution was determined using inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7500C). The relaxation time T1 and T1-weighted images were obtained using a MRI Analyzing and Imaging system Micro MR20-025H-I with a 0.47 T magnet (Shanghai Niumag Corporation). MRI in vivo was performed with MesoMR-60 MRI system with a 0.50 T magnet (Shanghai Niumag Corporation).Results and discussion
The prefabricated MnS aggregations were prepared by a simple solvothermal method. These aggregations were analyzed, using powder XRD, to identify the phase and crystallization. Fig. 1a shows that all the XRD peaks are in good agreement with crystalline MnS (JCPDS card 06-0518) with a cubic structure [space group: Fm3m(225)]. No characteristic peaks were observed for impurities, such as MnO, manganese dioxide and manganese(II,III) oxide (Mn3O4). The crystal cell parameters were calculated as a = 5.224 nm, b = 5.224 nm and c = 5.224 nm using JADE 5 software. These calculated values agreed with those in the JCPDS card 06-0518 file for MnS (a = 5.223 nm; b = 5.223 nm; c = 5.223 nm). Fig. 1b is a typical TEM image of the MnS aggregations. This image shows well dispersed spherical aggregations with an average size of 90 nm composed of ultrasmall nanocrystals. Fig. 1c shows the SAED pattern of the MnS aggregations with a crystalline nature. The cubic MnS nanocrystals obtained were well dispersed in cyclohexane with an obvious Tyndall effect as shown in Fig. 1d. The HRTEM image (Fig. 1d inset) indicates the presence of aggregations of small component particles of around 5 nm. The lattice spacing between the two adjacent planes was measured and found to be approximately 0.18 nm, which corresponded to the (220) lattice plane in cubic MnS. The EDX spectrum in Fig. 1f illustrates Mn and S element signals with the correct ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.07. The reason for the formation of MnS aggregation is that the small hydroxyl groups of water (H2O) generated by TAA decompose and adsorb onto the MnS nanocrystals, which can interact with each other and result in a linkage of the neighboring particles.49,56–58
:1.07. The reason for the formation of MnS aggregation is that the small hydroxyl groups of water (H2O) generated by TAA decompose and adsorb onto the MnS nanocrystals, which can interact with each other and result in a linkage of the neighboring particles.49,56–58
 | ||
| Fig. 1 XRD pattern (a), TEM image (b), SAED pattern (c), digital photograph image (in cyclohexane) (d), HRTEM image (e) and EDX spectrum (f) of the prepared MnS aggregations. The inset of (e) is the HRTEM image under low magnification. | ||
Prefabricated MnS aggregations that were dispersed in cyclohexane were added to an aqueous of sodium citrate with shaking. After standing for 10 min, the MnS could be transferred into the aqueous phase. It was surprising that the MnS nanocrystals that were dispersed in water depolymerized to small nanocrystals of approximately 5 nm, as observed by TEM and as shown in Fig. 2a. The SAED pattern of the sample (Fig. 2c) clearly indicated that it was crystalline MnS. The MnS nanocrystals produced in aqueous solution were further characterized using HRTEM, which showed a plane spacing of approximately 0.27 nm corresponding to the (200) planes of the cubic MnS. Fig. 2f shows the EDX spectrum of the MnS nanocrystals, and the elements of sulfur and manganese indicate that the phase transfer reaction did not influence the elementary composition of MnS. The phase transfer progress is shown schematically in Fig. 3. Oleic acid molecules that adsorbed onto the MnS nanocrystal surface were instead adsorbed by citrate ions with a short chain because of their stronger binding in water. As a result, the MnS nanocrystal aqueous solution can be stored for several months without forming a precipitate, which demonstrates the excellent colloidal stability.
 | ||
| Fig. 2 TEM image (a), size distribution (b), SAED pattern (c), digital photograph image (in H2O) (d) HRTEM image (e), and EDX spectrum (f) of the prepared MnS ultrasmall nanocrystals. | ||
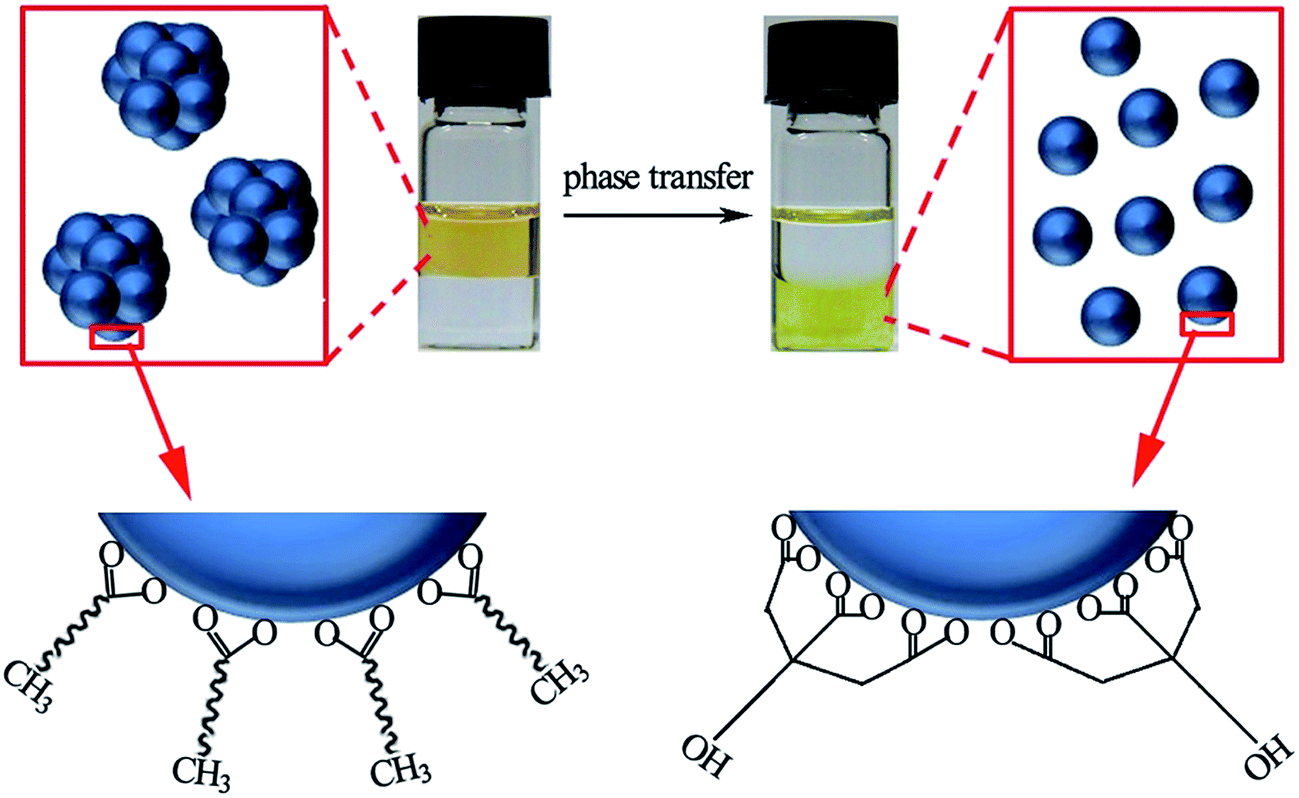 | ||
| Fig. 3 Photograph and schematic images of the phase transfer from cyclohexane to aqueous sodium citrate. | ||
The surface molecules of MnS nanocrystals before and after phase transfer were studied using FTIR. Fig. 4a shows the FTIR spectrum of the MnS nanocrystals before phase transfer, in which the 624 cm−1 bands refer to the Mn–S vibration of MnS.59,60 The broad band at approximately 3422 cm−1 was assigned to the stretching vibrations of the O–H bonds in the adsorbed water molecules, and the absorption at approximately 2925 cm−1 corresponded to the vibrations of the C–H bonds. Peaks at approximately 1640 and 1411 cm−1 are also found in the spectrum and were assigned to the asymmetric and symmetric stretching of the carboxylic group of the bound oleic acid.61–63 After the phase transfer, the FTIR spectrum of MnS nanocrystals in Fig. 4b shows the same band at 624 cm−1 as that before the phase transfer, indicating the existence of MnS. In contrast, the C–H bonds at around 2900 cm−1 were almost dispersed, and this was caused by the fewer C–H bonds found in Na3Cit than in oleic acid. Furthermore, the peaks of a carboxylic group at approximately 1622 cm−1 and 1399 cm−1 were much more intense than those before the phase transfer, resulting from the many more carboxylic groups found in Na3Cit than in oleic acid molecules. Therefore, these results confirm that Na3Cit molecules replaced the oleic acid on the MnS nanocrystal surface during the phase transfer.
 | ||
| Fig. 4 FTIR of MnS aggregations (a) and MnS nanocrystals that were coated with citrate groups (b). | ||
The prepared monodispersed MnS nanocrystals in aqueous solution were investigated as potential MRI T1 contrast agents. To characterize their relaxation efficiency, the Mn2+ concentration in the solution was determined using ICP-MS. Solutions with different Mn2+ concentrations diluted with hyperpure water were then analysed using the 0.47 T MRI Analyzing and Imaging system. Fig. 5a shows the relaxation rates 1/T1 as a function of the Mn2+ concentration for the MnS nanocrystals, in which the T1 relaxation processes result from the interaction between the excited nuclei and their surrounding environment. The r1 is approximately 3.4 s−1 mM−1 from the slope of the plot of 1/T1 versus the Mn2+ concentration. The r1 value of 3.4 s−1 mM−1 at 0.47 T for the ultrasmall nanocrystals was higher than that reported for Mn-based contrast agents measured under the same magnetic field. The value was even higher than the most reported values for Mn-based contrast agents tested in a high magnetic field (i.e., 1.5 T, 3.0 T, 7.0 T and 11.7 T), as shown in Table 1. Although two of the MnO nanocrystal contrast agents listed in Table 1 were higher than the MnS nanocrystals, the magnetic field strength that was applied (1.5 T) was much higher than that used in this experimental work (0.47 T). The high relaxivity in this research was attributed to their ultrasmall size and short chain capping molecules. The prepared MnS nanocrystals, therefore, indicated a promising potential for the use as MRI T1 contrast agents. T1-weighted MR imaging was further conducted to verify the potential use of MnS nanocrystals as MRI contrast agents. The concentration dependent brightening T1 effects which were because of the presence of MnS samples are presented in Fig. 5c. The T1 contrast enhancement effects were observed as the increased metal (Mn) concentration in the weighted image.
 | ||
| Fig. 5 Relaxivity with different Mn2+ concentrations for the MnS nanocrystal contrast agent in hyperpure H2O (a) and BSA solution (b) at 0.47 T; corresponding to T1-weighted images in hyperpure H2O (c) and BSA solution (d). | ||
| Sample composition | Material | Surface compound | Diameter (nm) | r1 (mM−1 s−1) | B0 (T) | Ref. |
|---|---|---|---|---|---|---|
| a Au@MnO = gold@manganese oxide, HAS-MONP = human albumin coated manganese oxide nanoparticles, HMnO@mSiO2 = mesoporous silica coated hollow manganese oxide nanoparticles, HMONs = hollow manganese oxide nanoparticles, Ir = iridium, PEG = poly(ethylene) glycol, PSS = poly(styrene) sulfate. | ||||||
| MnO | MnO | PEG–phospholipid | 7 | 0.37 | 3.0 | 13 |
| 15 | 0.18 | 13 | ||||
| 20 | 0.13 | 13 | ||||
| 25 | 0.12 | 13 | ||||
| HAS-MONP | MnO | ∼20 | 1.97 | 7.0 | 67 | |
| HMnO@mSiO2 | MnO | SiO2 | 65 | 1.72 | 1.5 | 32 |
| MnO | MnO | D-Glucuronic acid | 2.5 | 7.02 | 1.5 | 68 |
| MnO nanoplates | MnO | Amine-terminated PEG | 8 | 5.5 | 1.5 | 15 |
| MnO | MnO | Dopamine–human serum albumin | 40 | 1.9 | 7.0 | 43 |
| HMnO | MnO | PEG–phospholipid | 20 | 1.1 | 1.5 | 69 |
| HMnO | MnO | mSiO2 | 65 | 1 | 11.7 | 32 |
| HMnO | MnO | mSiO2(Ir)PEG | 80 | 0.2 | 0.47 | 70 |
| Au@MnO | MnO | Fluorescent PEG | 18 | 0.2 | 3.0 | 71 |
| Spherical Mn3O4 | Mn3O4 | 3 | 2.38 | 1.5 | 72 | |
| HMONs | Mn3O4 | 20 | 1.42 | 3.0 | 73 | |
| Mn3O4 nanospheres | Mn3O4 | (PSS) | 9.8 | 1.31 | 3.0 | 34 |
| Mn3O4 nanoplates | Mn3O4 | PSS | 10 | 2.06 | 34 | |
| Mn3O4 nanocubes | Mn3O4 | PSS | 5 | 1.08 | 34 | |
| Mn3O4 nanoplates | Mn3O4 | PEG–phospholipid | 9 | 0.1 | 1.5 | 74 |
| Mn3O4 | Mn3O4 | SiO2 conjugated with a fluorophore and folate | 35 | 0.5 | 3.0 | 75 |
In clinical situations, the agent reaches the target site be being transported in the plasma. Thus, it is important to evaluate the contrast effect of the MnS nanocrystals in the bloodstream.64 In this research, the prepared water-soluble MnS nanocrystals were dispersed in an aqueous solution of BSA for further T1 relaxivity testing because serum albumin is the richest protein of human blood plasma, playing a crucial role in the uptake, transportation, biodistribution and excretion of the contrast agent.65,66 The T1 relaxation rate (1/T1) of the MnS agent of the BSA solution (0.725 mmol L−1) is shown in Fig. 5b. The value of r1 is 8.7 s−1 mM−1, which is 2.6-fold higher than that in water. Fig. 5d shows five sets of the T1-weighted MR images of BSA containing MnS nanocrystals. The results suggest that the presence of BSA with the MnS contrast agent solution could provide an additional enhancement effect of T1 relaxivity. This enhancement agrees with the results found in the literature, and is caused by the BSA increasing the rotational correlation time (τ) of the nanocrystals in solution.
The toxicity of nanocrystals is also a critical issue when they are used as MRI contrast agents. In this research, the cell viabilities of the MnS nanocrystals were determined using a 12 h incubation of Hep G2 cells with the nanocrystals. As shown in Fig. 6, there was no obvious decrease in cell viability after the cells were incubated with MnS nanocrystals at different concentrations for 12 h. The viability of untreated cells was assumed to be 100%. The cell viability still remained as high as 95% after 12 h incubation at a concentration of 70 μg mL−1 Mn, indicating that the colloidally stable MnS nanocrystals have low toxicity and are suitable as MRI contrast agents within the previously mentioned nanocrystal concentration.
 | ||
| Fig. 6 In vitro cell cytotoxicity of Hep G2 cells that were incubated with MnS nanocrystals at different concentrations for 12 h. | ||
In vivo MRI of a nude mouse was performed at 0.47 T. The MnS nanocrystals were suspended in aqueous phase and then injected into the tail vein of a mouse at a dosage of 12.5 μmol kg−1 body weight. Note that the administered concentration of Mn in the dose of 12.5 μmol kg−1 is only 1/8–1/16 of the standard clinical dose of Gd–DTPA (0.1–0.2 mmol kg−1).56 Dynamic contrast enhanced T1-weighted images of the liver and kidney were obtained from the in vivo test.76,77 As shown in Fig. 7a, the T1-weighted MR images clearly show a slight contrast enhancement of the liver 10 min after injecting the MnS nanocrystals, and the image signals show the highest contrast enhancement 1 h post-injection. Such a significant change in contrast was attributed to the accumulation of the injected MnS nanocrystals in the liver. Then, 4 h post-injection, these hyperintense signals persisted, indicating that the agent in liver had a relatively longer residence time.
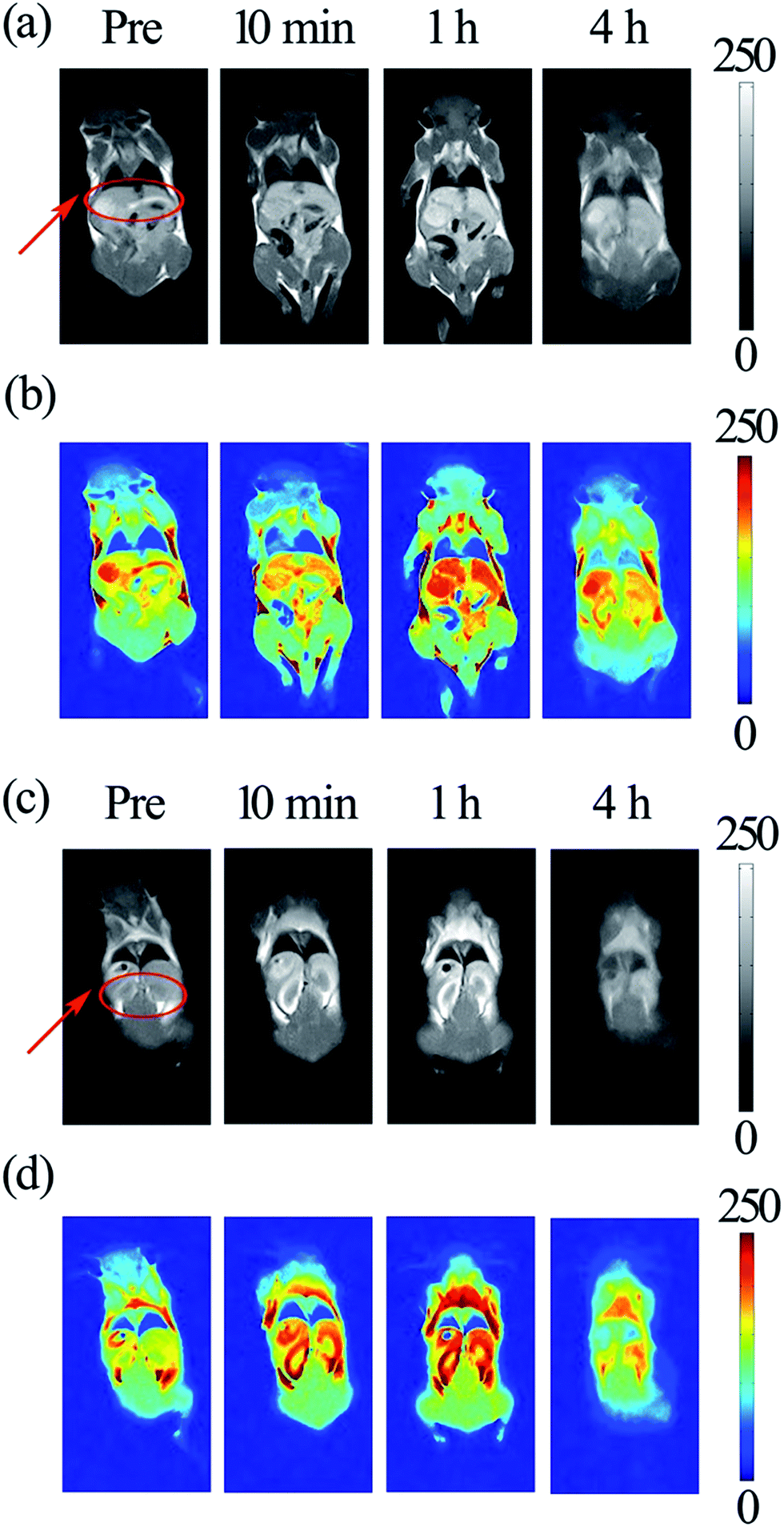 | ||
| Fig. 7 T1-weighted in vivo images obtained using the MnS nanocrystal contrast agent at 0.47 T: coronal gray views of liver (a) and kidney (c) as indicated with arrows at different times (as noted) after injecting 0.1 mL of aqueous sample into a mouse. Images (b) and (d) are pseudocolored images corresponding to (a) and (c), respectively. | ||
In addition, the corresponding kidney T1-weighted MRI are shown in Fig. 7c and shows a slight contrast agent enhancement in the kidney 10 min after injecting the MnS nanocrystals, and the image signals show a high contrast enhancement 1 h post-injection because of the accumulation of the MnS nanocrystal agent in the kidney. These hyperintense signals disappeared gradually, and the less hyperintense signals could be detected in the kidney 4 h post-injection. The corresponding pseudocolored images were provided to better demonstrate the high signal intensity of the mouse in the liver (Fig. 7b) and kidney (Fig. 7d).
To further investigate the metabolizable process of the prepared MnS nanocrystal contrast agent in the mouse body, a simplified model of the uptake and the excretion of the agent was followed using the method in reference.78 In this model, the signal intensity is considered to represent the uptake (distribution) and excretion (elimination) of the contrast agent, although the uptake and excretion of the contrast agent are actually very complex processes. To obtain dependable results for signal intensity measurement, ROIs of the kidney and liver were manually drawn on T1-weighted images, as shown in Fig. 8a. The data are shown to fit well to the model. According to the quantification analysis of the MR signal in the liver and kidney, there was a sharp increase (32%) in T1-weighted signal intensity 1 h post-injection in the liver and an increase of 30% in the kidney.
 | ||
| Fig. 8 Time dependence of the percentage enhancement in the kidney and liver after the intravenous administration of the MnS contrast agent (a). Time dependence of the contrast agent induced hepatic (b) and renal (c) intensity enhancement. The data points were obtained from the T1-weighted images, and the curves are fitted according to eqn (3) as described in the experimental section. | ||
The fitting curves of the MnS contrast agent induced hepatic and renal intensity enhancement are shown in Fig. 8b and c, respectively, and the corresponding parameters are listed in Table 2. The parameters in Table 2 show that the hepatic uptake rate of MnS is t1/2,uptake is 55 min, and the hepatic excretion rate is t1/2,excretion = 349 min. The renal uptake rate of MnS is t1/2,uptake = 38 min, and the renal excretion rate is t1/2,excretion = 159 min. Compared to the commercial T1 agent, Gd–DTPA, the prepared MnS nanocrystal contrast agent used in this work has a similar uptake rate and a lower excretion rate in the kidney and liver.66 The low excretion rate of the agent is accompanied by persistent MRI intensity enhancement of the contrast during the imaging period. The signal IE disappeared gradually in the kidney, implying that the MnS agent was removed from the blood and eliminated through the kidney. In addition to kidney elimination, the liver decay after injection probably supports the theory that the partial MnS nanocrystals are also being excreted through the hepatocytes into the bile and feces.79 Therefore, the large IEmax, quick uptake rate and slow excretion rate make the MnS nanocrystals produced in this research a good candidate for a contrast agent to provide intense and stable imaging contrast on a longer time scale with low toxicity.
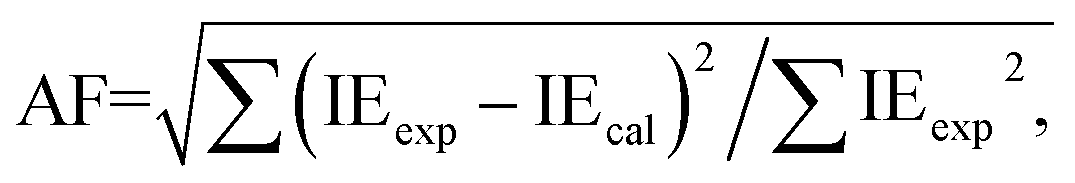 where IEexp and IEcal are the experimental and calculated intensity enhancements, respectively
where IEexp and IEcal are the experimental and calculated intensity enhancements, respectively
| Organ | IEmax | t1/2,uptake (min) | t1/2,excretion (min) | AF |
|---|---|---|---|---|
| Liver | 48 | 55 | 349 | 0.23 |
| Kidney | 45 | 38 | 159 | 0.22 |
Conclusion
In summary, smart MnS nanocrystals capped with citrate were prepared using the easy phase transfer method. XRD characterization indicated that the MnS nanocrystals had a stable cubic crystalline structure. The MnS nanocrystals were ultrasmall in size (approximately 5 nm), with high monodispersion in aqueous solution as observed under TEM. The cell viabilities of up to 95% when incubated with Hep G2 cells demonstrated the low toxicity of the MnS sample prepared. As potential T1 MRI contrast agents, the MnS nanocrystals showed a high relaxivity of 3.4 s−1 mM−1 in a 0.47 T magnetic field. The relaxivity reached 8.7 s−1 mM−1 when tested in aqueous BSA. The nanocrystals were further applied to the MRI contrast agent in vivo at a low dose, resulting in the obvious contrast enhancement for the liver and kidney in the mouse. The metabolic study displayed the quick uptake rate and slow excretion rate for the MnS nanocrystals in the mouse body. These results suggest that the prepared smart MnS nanocrystal agents are strong candidate T1 contrast agents for the next generation of MRIs.Acknowledgements
This study is supported by National Science Foundation of China (51402201); the Beijing Youth Excellent Talent Program (CIT&TCD201404162); the Beijing Local College Innovation Team Improve Plan (IDHT20140512); and the Scientific Research Base Development Program of the Beijing Municipal Commission of Education.References
- O. Chen, L. Riedemann, F. Etoc, H. Herrmann, M. Coppey, M. Barch, C. T. Farrar, J. Zhao, O. T. Bruns and H. Wei, Nat. Commun., 2014, 5, 5093 CrossRef CAS PubMed.
- E. Terreno, D. D. Castelli, A. Viale and S. Aime, Chem. Rev., 2010, 110, 3019–3042 CrossRef CAS PubMed.
- T. Macher, J. Totenhagen, J. Sherwood, Y. Qin, D. Gurler, M. S. Bolding and Y. Bao, Adv. Funct. Mater., 2015, 25, 490–494 CrossRef CAS.
- K. Yang, G. Yang, L. Chen, L. Cheng, L. Wang, C. Ge and Z. Liu, Biomaterials, 2015, 38, 1–9 CrossRef CAS PubMed.
- J. Zhang, Y. Ye, Y. Chen, C. Pregot, T. Li, S. Balasubramaniam, D. B. Hobart, Y. Zhang, S. Wi and R. M. Davis, J. Am. Chem. Soc., 2014, 136, 2630–2636 CrossRef CAS PubMed.
- Q. Tian, J. Hu, Y. Zhu, R. Zou, Z. Chen, S. Yang, R. Li, Q. Su, Y. Han and X. Liu, J. Am. Chem. Soc., 2013, 135, 8571–8577 CrossRef CAS PubMed.
- X. Zhu, J. Zhou, M. Chen, M. Shi, W. Feng and F. Li, Biomaterials, 2012, 33, 4618–4627 CrossRef CAS PubMed.
- H. Yang, Y. Zhuang, Y. Sun, A. Dai, X. Shi, D. Wu, F. Li, H. Hu and S. Yang, Biomaterials, 2011, 32, 4584–4593 CrossRef CAS PubMed.
- L. Wang, X. Zhu, X. Tang, C. Wu, Z. Zhou, C. Sun, S.-L. Deng, H. Ai and J. Gao, Chem. Commun., 2015, 51, 4390–4393 RSC.
- G. Huang, J. Hu, H. Zhang, Z. Zhou, X. Chi and J. Gao, Nanoscale, 2014, 6, 726–730 RSC.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- J. W. M. Bulte and D. L. Kraitchman, NMR Biomed., 2004, 17, 484–499 CrossRef CAS PubMed.
- H. B. Na, J. H. Lee, K. An, Y. I. Park, M. Park, I. S. Lee, D.-H. Nam, S. T. Kim, S.-H. Kim, S.-W. Kim, K.-H. Lim, K.-S. Kim, S.-O. Kim and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 5397–5401 CrossRef CAS PubMed.
- F. Chen, W. Bu, S. Zhang, X. Liu, J. Liu, H. Xing, Q. Xiao, L. Zhou, W. Peng and L. Wang, Adv. Funct. Mater., 2011, 21, 4285–4294 CrossRef CAS.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. B. Na, J.-G. Park, T.-Y. Ahn, Y.-W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624–12631 CrossRef CAS PubMed.
- P. H. Kuo, E. Kanal, A. K. Abu-Alfa and S. E. Cowper, Radiology, 2007, 242, 647–649 CrossRef PubMed.
- T. A. Rose and J. W. Choi, Am. J. Med., 2015, 128, 943–949 CrossRef PubMed.
- R. Chen, D. Ling, L. Zhao, S. Wang, Y. Liu, R. Bai, S. Baik, Y. Zhao, C. Chen and T. Hyeon, ACS Nano, 2015, 9, 12425–12435 CrossRef CAS PubMed.
- M. Park, N. Lee, S. H. Choi, K. An, S.-H. Yu, J. H. Kim, S.-H. Kwon, D. Kim, H. Kim, S.-I. Baek, T.-Y. Ahn, O. K. Park, J. S. Son, Y.-E. Sung, Y.-W. Kim, Z. Wang, N. Pinna and T. Hyeon, Chem. Mater., 2011, 23, 3318–3324 CrossRef CAS.
- U. I. Tromsdorf, O. T. Bruns, S. C. Salmen, U. Beisiegel and H. Weller, Nano Lett., 2009, 9, 4434–4440 CrossRef CAS PubMed.
- L. Sandiford, A. Phinikaridou, A. Protti, L. K. Meszaros, X. Cui, Y. Yan, G. Frodsham, P. A. Williamson, N. Gaddum and R. M. Botnar, ACS Nano, 2012, 7, 500–512 CrossRef PubMed.
- F. Hu, Q. Jia, Y. Li and M. Gao, Nanotechnology, 2011, 22, 245604 CrossRef PubMed.
- V. K. Sharma, A. Alipour, Z. Soran-Erdem, Z. G. Aykut and H. V. Demir, Nanoscale, 2015, 7, 10519–10526 RSC.
- M. Kueny-Stotz, A. Garofalo and D. Felder-Flesch, Eur. J. Inorg. Chem., 2012, 2012, 1987–2005 CrossRef CAS.
- X. Yu, Y. Z. Wadghiri, D. H. Sanes and D. H. Turnbull, Nat. Neurosci., 2005, 8, 961–968 CrossRef CAS PubMed.
- S. L. O'Neal and W. Zheng, Curr Environ Health Rep, 2015, 2, 315–328 CrossRef PubMed.
- M. E. Bernardino, J. C. Weinreb, D. G. Mitchell, W. C. Small and M. Morris, Magn. Reson. Imaging, 1994, 4, 872–876 CrossRef CAS.
- B. Y. W. Hsu, M. Ng, Y. Zhang, S. Y. Wong, K. Bhakoo, X. Li and J. Wang, Adv. Funct. Mater., 2015, 25, 5269–5276 CrossRef CAS.
- C. R. Gordijo, A. Z. Abbasi, M. A. Amini, H. Y. Lip, A. Maeda, P. Cai, P. J. O'Brien, R. S. DaCosta, A. M. Rauth and X. Y. Wu, Adv. Funct. Mater., 2015, 25, 1858–1872 CrossRef CAS.
- Z. Zhen and J. Xie, Theranostics, 2012, 2, 45 CrossRef CAS PubMed.
- X. Yang, Z. Zhou, L. Wang, C. Tang, H. Yang and S. Yang, Mater. Res. Bull., 2014, 57, 97–102 CrossRef CAS.
- T. Kim, E. Momin, J. Choi, K. Yuan, H. Zaidi, J. Kim, M. Park, N. Lee, M. T. McMahon and A. Quinones-Hinojosa, J. Am. Chem. Soc., 2011, 133, 2955–2961 CrossRef CAS PubMed.
- Y. Chen, Q. Yin, X. Ji, S. Zhang, H. Chen, Y. Zheng, Y. Sun, H. Qu, Z. Wang and Y. Li, Biomaterials, 2012, 33, 7126–7137 CrossRef CAS PubMed.
- C.-C. Huang, N.-H. Khu and C.-S. Yeh, Biomaterials, 2010, 31, 4073–4078 CrossRef CAS PubMed.
- F. Hu and Y. S. Zhao, Nanoscale, 2012, 4, 6235–6243 RSC.
- X. Yang, Y. Wang, Y. Sui, X. Huang, T. Cui, C. Wang, B. Liu, G. Zou and B. Zou, Langmuir, 2012, 28, 17811–17816 CrossRef CAS PubMed.
- S. W. Kennedy, K. Harris and E. Summerville, J. Solid State Chem., 1980, 31, 355–359 CrossRef CAS.
- C. Sombuthawee, S. B. Bonsall and F. A. Hummel, J. Solid State Chem., 1978, 25, 391–399 CrossRef CAS.
- J. Lu, P. Qi, Y. Peng, Z. Meng, Z. Yang, W. Yu and Y. Qian, Chem. Mater., 2001, 13, 2169–2172 CrossRef CAS.
- H. M. Chilton, S. C. Jackels, W. H. Hinson and K. E. Ekstrand, J. Nucl. Med., 1984, 25, 604–607 CAS.
- G. K. Das, N. J. J. Johnson, J. Cramen, B. Blasiak, P. Latta, B. Tomanek and F. C. J. M. van Veggel, J. Phys. Chem. Lett., 2012, 3, 524–529 CrossRef CAS PubMed.
- R. Xing, F. Zhang, J. Xie, M. Aronova, G. Zhang, N. Guo, X. Huang, X. Sun, G. Liu and L. H. Bryant, Nanoscale, 2011, 3, 4943–4945 RSC.
- J. Huang, J. Xie, K. Chen, L. Bu, S. Lee, Z. Cheng, X. Li and X. Chen, Chem. Commun., 2010, 46, 6684–6686 RSC.
- M. Elsabahy, G. S. Heo, S.-M. Lim, G. Sun and K. L. Wooley, Chem. Rev., 2015, 115, 10967–11011 CrossRef CAS PubMed.
- D. Ling, N. Lee and T. Hyeon, Acc. Chem. Res., 2015, 48, 1276–1285 CrossRef CAS PubMed.
- X. Cao, F. Cao, L. Xiong, Y. Yang, T. Cao, X. Cai, W. Hai, B. Li, Y. Guo and Y. Zhang, Nanoscale, 2015, 7, 13404–13409 RSC.
- E. Peng, F. Wang, B. Zheng, S. F. Y. Li and J. M. Xue, Nanoscale, 2015, 7, 7819–7832 RSC.
- F. Zhang, E. Lees, F. Amin, P. Rivera_Gil, F. Yang, P. Mulvaney and W. J. Parak, Small, 2011, 7, 3113–3127 CrossRef CAS PubMed.
- J. Liu, Z. Sun, Y. Deng, Y. Zou, C. Li, X. Guo, L. Xiong, Y. Gao, F. Li and D. Zhao, Angew. Chem., Int. Ed., 2009, 48, 5875–5879 CrossRef CAS PubMed.
- L. An, H. Hu, J. Du, J. Wei, L. Wang, H. Yang, D. Wu, H. Shi, F. Li and S. Yang, Biomaterials, 2014, 35, 5381–5392 CrossRef CAS PubMed.
- X. Liang, Y. Li, X. Li, L. Jing, Z. Deng, X. Yue, C. Li and Z. Dai, Adv. Funct. Mater., 2015, 25, 1451–1462 CrossRef CAS.
- T. Passuello, M. Pedroni, F. Piccinelli, S. Polizzi, P. Marzola, S. Tambalo, G. Conti, D. Benati, F. Vetrone and M. Bettinelli, Nanoscale, 2012, 4, 7682–7689 RSC.
- M. P. Leal, S. Rivera-Fernández, J. M. Franco, D. Pozo, M. Jesús and M. L. García-Martín, Nanoscale, 2015, 7, 2050–2059 RSC.
- Z. Chen, H. Chen, H. Hu, M. Yu, F. Li, Q. Zhang, Z. Zhou, T. Yi and C. Huang, J. Am. Chem. Soc., 2008, 130, 3023–3029 CrossRef CAS PubMed.
- E. Peng, J. Ding and J. M. Xue, J. Mater. Chem., 2012, 22, 13832–13840 RSC.
- H. J. Weinmann, R. C. Brasch, W. R. Press and G. E. Wesbey, Am. J. Roentgenol., 1984, 142, 619–624 CrossRef CAS PubMed.
- S. Xu, Z.-H. Li, Q. Wang, L.-J. Cao, T.-M. He and G.-T. Zou, J. Alloys Compd., 2008, 465, 56–60 CrossRef CAS.
- H.-B. Xia, J. Yi, P.-S. Foo and B. Liu, Chem. Mater., 2007, 19, 4087–4091 CrossRef CAS.
- H. Yang, J. Zhao, L. Song, L. Shen, Z. Wang, L. Wang and D. Zhang, Mater. Lett., 2003, 57, 2287–2291 CrossRef CAS.
- T. Dhandayuthapani, M. Girish, R. Sivakumar, C. Sanjeeviraja and R. Gopalakrishnan, Appl. Surf. Sci., 2015, 353, 449–458 CrossRef CAS.
- Y. Tian, H. Y. Yang, S. Yu and X. Li, ChemPlusChem, 2014, 79, 1584–1589 CrossRef CAS.
- N. Shukla, C. Liu, P. M. Jones and D. Weller, J. Magn. Magn. Mater., 2003, 266, 178–184 CrossRef CAS.
- L. Zhang, R. He and H.-C. Gu, Appl. Surf. Sci., 2006, 253, 2611–2617 CrossRef CAS.
- Q. Mu, G. Jiang, L. Chen, H. Zhou, D. Fourches, A. Tropsha and B. Yan, Chem. Rev., 2014, 114, 7740–7781 CrossRef CAS PubMed.
- J. Li, P. Cai, A. Shalviri, J. T. Henderson, C. He, W. D. Foltz, P. Prasad, P. M. Brodersen, Y. Chen and R. DaCosta, ACS Nano, 2014, 8, 9925–9940 CrossRef CAS PubMed.
- Z. Li, W. Li, X. Li, F. Pei, Y. Li and H. Lei, Magn. Reson. Imaging, 2007, 25, 412–417 CrossRef CAS PubMed.
- T. D. Schladt, K. Schneider, M. I. Shukoor, F. Natalio, H. Bauer, M. N. Tahir, S. Weber, L. M. Schreiber, H. C. Schroder, W. E. G. Muller and W. Tremel, J. Mater. Chem., 2010, 20, 8297–8304 RSC.
- M. J. Baek, J. Y. Park, W. Xu, K. Kattel, H. G. Kim, E. J. Lee, A. K. Patel, J. J. Lee, Y. Chang, T. J. Kim, J. E. Bae, K. S. Chae and G. H. Lee, ACS Appl. Mater. Interfaces, 2010, 2, 2949–2955 CAS.
- K. An, S. G. Kwon, M. Park, H. B. Na, S.-I. Baik, J. H. Yu, D. Kim, J. S. Son, Y. W. Kim, I. C. Song, W. K. Moon, H. M. Park and T. Hyeon, Nano Lett., 2008, 8, 4252–4258 CrossRef CAS PubMed.
- Y.-K. Peng, C.-W. Lai, C.-L. Liu, H.-C. Chen, Y.-H. Hsiao, W.-L. Liu, K.-C. Tang, Y. Chi, J.-K. Hsiao, K.-E. Lim, H.-E. Liao, J.-J. Shyue and P.-T. Chou, ACS Nano, 2011, 5, 4177–4187 CrossRef CAS PubMed.
- T. D. Schladt, M. I. Shukoor, K. Schneider, M. N. Tahir, F. Natalio, I. Ament, J. Becker, F. D. Jochum, S. Weber, O. Köhler, P. Theato, L. M. Schreiber, C. Sönnichsen, H. C. Schröder, W. E. G. Müller and W. Tremel, Angew. Chem., Int. Ed., 2010, 49, 3976–3980 CrossRef CAS PubMed.
- K. An, M. Park, J. H. Yu, H. B. Na, N. Lee, J. Park, S. H. Choi, I. C. Song, W. K. Moon and T. Hyeon, Eur. J. Inorg. Chem., 2012, 2012, 2148–2155 CrossRef CAS.
- J. Shin, R. M. Anisur, M. K. Ko, G. H. Im, J. H. Lee and I. S. Lee, Angew. Chem., Int. Ed., 2009, 48, 321–324 CrossRef CAS PubMed.
- T. Yu, J. Moon, J. Park, Y. I. Park, H. B. Na, B. H. Kim, I. C. Song, W. K. Moon and T. Hyeon, Chem. Mater., 2009, 21, 2272–2279 CrossRef CAS.
- H. Yang, Y. Zhuang, H. Hu, X. Du, C. Zhang, X. Shi, H. Wu and S. Yang, Adv. Funct. Mater., 2010, 20, 1733–1741 CrossRef CAS.
- X. Zhu, X. Chi, J. Chen, L. Wang, X. Wang, Z. Chen and J. Gao, Anal. Chem., 2015, 87, 8941–8948 CrossRef CAS PubMed.
- Z. Zhao, X. Wang, Z. Zhang, H. Zhang, H. Liu, X. Zhu, H. Li, X. Chi, Z. Yin and J. Gao, ACS Nano, 2015, 9, 2749–2759 CrossRef CAS PubMed.
- J. Feng, G. Sun, F. Pei and M. Liu, J. Inorg. Biochem., 2002, 92, 193–199 CrossRef CAS PubMed.
- J. Yu, W. Yin, X. Zheng, G. Tian, X. Zhang, T. Bao, X. Dong, Z. Wang, Z. Gu and X. Ma, Theranostics, 2015, 5, 931–945 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra24775f |
| This journal is © The Royal Society of Chemistry 2016 |