
Peculiarities of 2-amino-3-R-4-aryl-4H-pyranes multicomponent synthesis derived from 1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide†
Dmitry A. Legaa,
Nikolay Yu. Gorobetsb,
Valentine P. Chernykha,
Svetlana V. Shishkinab and
Leonid A. Shemchuk*a
aNational University of Pharmacy, Pushkinska Str. 53, Kharkiv, 61001, Ukraine. E-mail: dr_shemchuk@mail.ru
bSSI ‘Institute for Single Crystals’ of NAS of Ukraine, Lenin Ave. 60, Kharkiv, 61001, Ukraine
First published on 29th January 2016
Abstract
The new 2-amino-3-R-4-aryl-6-ethyl-4,6-dihydropyrano[3,2-c][2,1]benzothiazine 5,5-dioxides were synthesized via three-component interaction of 1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide with arylcarbaldehydes and active methylene nitriles. Depending on the nature of an active methylene nitrile and an arylcarbaldehyde this interaction can lead either to the target 2-amino-4H-pyrans or to the stable triethylammonium salts of bis(1H-2,1-benzothiazin-4(3H)-one 2,2-dioxides) (bis-adducts). The latter is a completely new product of such interaction. The structure of the bis-adduct was confirmed by single crystal X-ray diffraction. Actually, the formation of stable triethylammonium salts (as the process competitive with the 2-amino-4H-pyrans formation) appeared to be reversible and their interaction with active methylene nitriles led to the formation of 2-amino-4H-pyrans. The extended and adjusted mechanism of the three-component interaction, that includes the bis-adducts formation stage, was proposed. Taking into account the peculiarities of the mechanism, we were capable to control the reaction selectivity.
1. Introduction
Multicomponent reactions (MCRs) are an extremely effective tool for the rapid generation of small-molecule libraries by the creation of structural complexity in a single step from three or more reactants, providing greater efficiency and an atom economy as compared with stepwise synthesis.1,2 Therefore, there is a continuous growth of interest in MCRs investigation, especially for their possible application in combinatorial and medicinal science. In part, the pharmaceutical industry has fueled this resurgence because of the growing need to assemble libraries of structurally complex substances for evaluation as lead compounds for drug discovery and development.3 However, the multicomponent processes are often associated with ambiguous reaction mechanisms and selectivity issues or unexpected outcome4 making the design of novel MCRs as well as an extension of the scope of known MCRs to be an intriguing but challenging task.In our previous paper5 we described a three-component interaction of 1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide with active methylene nitriles and isatines. This interaction led to the formation of the corresponding fused 2-amino-4H-pyrans spirocondensed with 2-oxindol ring. This article was the first, dedicated to the multicomponent interactions of such benzothiazinones.
1H-2,1-Benzothiazin-4(3H)-one 2,2-dioxide represents the active methylene CH-acid. Its structure is an analogue of a cyclic active methylene 1,3-dicarbonyl compounds. This makes it a very convenient and promising synthon and opens great opportunities for building of a new heterocyclic systems based on CH2–CO fragment in its molecule. One of the most prospective routes to achieve this goal is using of MCRs. Unlike its carbonyl analogue, 1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide exists entirely in the 4-oxoform. Simultaneously, the carbonyl group of the given heterocycle is distinguished by a high propensity for enolization in the course of introducing an alkyl or an acyl groups into position 3.6,7
Furthermore, 1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide derivatives have recently gained an additional value due to their reported biological activities such as potent antibacterial effect,8 lipoxygenase inhibition and, so, their potential for heart diseases treatment.9 Moreover, 1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide core is bioisosteric to the 2,3-dihydro-4H-1,2-benzothiazin-4-one 1,1-dioxide (Fig. 1). The last one is a motif of well-known analgesic and anti-inflammatory agents (Piroxicam®, Droxicam® and Meloxicam® and its heteroanologues – Tenoxicam® and Lornoxicam®). Interestingly, isosteric benzothiazine-3-carboxamides, Fig. 2A(d) have been shown to posses much higher analgesic activity than Piroxicam® and Meloxicam®, Fig. 2A(a) and (b).10–12
 | ||
| Fig. 1 Common scaffold of applied analgesics (left side) and 1H-2,1-benzothiazin-4(3H)-one 2,2-dioxides possessing higher analgesic activity (right side). | ||
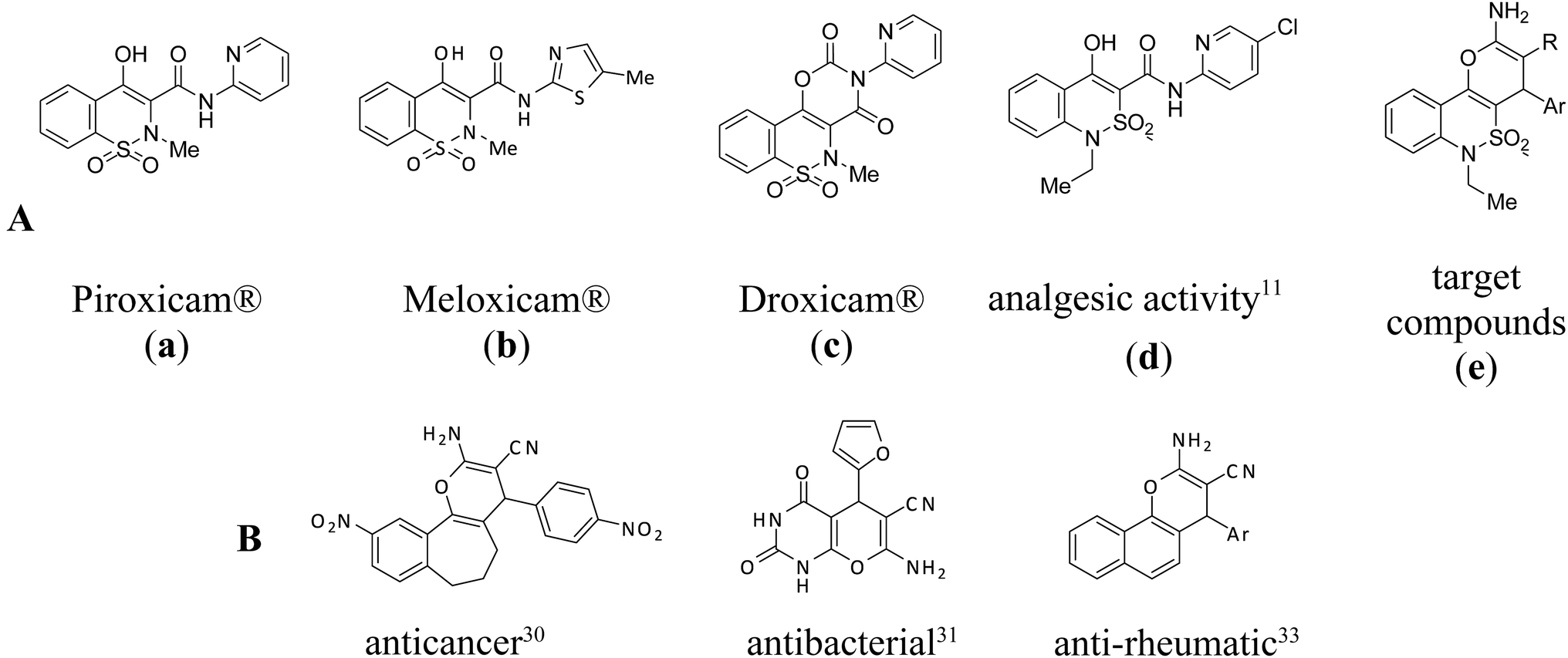 | ||
| Fig. 2 Examples of bioactive benzothiazines (A), 2-amino-3-cyano-4H-pyranes (B) and the target compounds (e). | ||
Multicomponent interactions of enol-nucleophiles (1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide is one of them) with carbonyl compounds and active methylene nitriles are attracting synthetic community for a long time. The reason is the formation of various products depending on the reaction conditions and structure of the starting compounds. In the most cases, such interaction is the direct route for the 2-amino-4H-pyran core construction. The main advantage of this methodology is the reduction of synthetic steps, which is of paramount importance for the preparation of intermediate unsaturated nitriles which are, in many cases, toxic and lachrymatory.13–15 To date, a lot of approaches were applied for three-component synthesis of 2-amino-4H-pyrans. Traditionally, the interactions of 1,3-dicarbonyl compounds with active methylene nitriles and benzaldehydes are easily carried out on heating in ethanol with basic catalysts, such as triethylamine,16,17 piperidine,18 morpholine,15 which resulted into formation of 2-amino-4H-pyrans in good to excellent yields.
In their turn, condensed 4H-pyranes are ones of the most well-known natural and synthetic heterocyclic compounds. They attract considerable attention since they possess an extensive range of biological effects. Several naturally occurring pyran-annulated heterocyclic compounds display anti-inflammatory,19 anti-HIV activity,20 cytotoxic activity against leukemia cells,21 antileishmanial activity,22 and slight activity against hepatitis B virus.23 Synthetically available 4H-pyranes are also used as pigments24 and photoactive materials.25 Furthermore, some derivatives of 4H-pyranes (for example cromakalim) can serve as a typical ATP-sensitive potassium channel openers or possess potent relaxant activity on blood vessels, cardiac muscle, and other smooth muscles. Thus, they may find an application in the treatment of variety of diseases such as hypertension, asthma, ischemia, and urinary incontinence.26,27 Moreover, several representatives of this class of compounds are known to be cognitive enhancers used to treat neurodegenerative disorders, including Alzheimer's, Huntington's, Parkinson's diseases and schizophrenia.28,29 In previous studies, 2-amino-3-cyano-4H-pyran derivatives, which are closely related to the title compounds, were found to possess anticancer,30 antibacterial,31,32 and anti-rheumatic33 effects; selected representatives of them are given in Fig. 2B.
In continuation of our previous researches, the reaction of 1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide with an active methylene nitriles and arylcarbaldehydes to synthesize corresponding 4-aryl-4H-pyran derivatives fused with 1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide, Fig. 2A(e), was investigated. It acquires additional value, because of, currently, 2,3-dihydro-4H-thiochromen-4-one is an example of six-membered sulfur containing heterocycles, based on which, 2-amino-4H-pyranes were obtained.15,34
2. Results and discussion
The first step of our researches was to find out the most suitable catalyst for three-component interaction of 1-ethyl-1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide with malononitrile and benzaldehyde. As representatives, we applied triethanolamine (used in our previous work5), triethylamine (as an inexpensive and widely used organic base) and urea (as an environmentally benign organo-catalyst and since it allows application of relatively mild reaction conditions13) as catalysts and ethanol or aqueous ethanol as solvents. However, the use of urea as a catalyst showed the low efficiency. The reactions with malononitrile resulted in the corresponding 2-amino-3-cyano-4-phenyl-4H-pyran in 30–45% yields, which were considered as unsatisfactory. In this regard, triethanolamine and triethylamine demonstrated much higher efficiency, since their applying in MCR allowed us to obtain condensed 2-amino-4-phenyl-4H-pyran in high yields. Thus, triethylamine was used in our further experiments.In general, the MCR between 1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide 1, malononitrile (2) and benzaldehydes 3a–g was carried out in refluxing ethanol during 1 hour in the presence of equimolar quantities of triethylamine and it resulted in the formation of the expected 2-amino-4H-pyran derivatives 5a–g isolated in high yields (Table 1, Method A). The products precipitated from the reaction mixture were recrystallized from ethanol. For 4-nitrobenzaldehyde (3b), the reaction temperature and time were decreased up to 50 °C for 40 min to avoid formation of undesirable side products. Though one could expect the reactivity decreases for 4-dimethylaminobenzaldehyde (3f) due to its strong electron donating substituent effect,35 or for bulky 9-anthraldehyde (3g), the high yields were also obtained in these cases. The application of the stepwise approach towards 2-amino-4H-pyranes 5a–g (Table 1, Method B) resulted in the lower yields compared to those, obtained by the MCRs. The gathered results confirm the preferred applicability of the multicomponent format for synthesis of 5a–g and serve as evidence that the formation of 2-amino-4H-pyran heterocycle in the course of MCR most likely proceeds via intermediate arylidenes 4.
|
|||
|---|---|---|---|
| Compound | Ar | Yieldsb, % | |
| Method A | Method B | ||
| a Reagents and conditions: (i) EtOH, Et3N (1.0 equiv.), reflux for 1 h (for 5b heating at 50 °C for 40 min in both methods).b Isolated yield. | |||
| 5a | C6H5 | 82 | 55 |
| 5b | 4-NO2-C6H4 | 95 | 71 |
| 5c | 2-MeO-C6H4 | 85 | 66 |
| 5d | 4-MeO-C6H4 | 84 | 58 |
| 5e | 4-Cl-C6H4 | 89 | 81 |
| 5f | 4-Me2N-C6H4 | 81 | 63 |
| 5g | 9-Anthracenyl | 93 | 91 |

The simple performance of the reaction given above, encouraged us to introduce other active methylene nitriles into this MCR. Ethyl cyanoacetate (6), benzylcyanide (12) and N-(p-tolyl)cyanoacetamide (13) were chosen as representative building blocks to investigate the scope and limitations the reaction.
However, for ethyl cyanoacetate (6) reacted under similar conditions during 4 hours, a significant decrease in the process efficiency and selectivity (Scheme 1) was observed. The only exception was the starting 4-nitrobenzaldehyde (3b) for which a pure target 2-amino-4H-pyran 7b was isolated in 51% yield. For aldehydes 3f,g the reaction stopped on the formation of arylidenes 9f,g. In spite of our attempts to prolong the reaction time or to use a higher boiling point solvent (DMF), we failed to obtain the expected products 7f,g.
 | ||
| Scheme 1 Reagents and conditions: (i) EtOH (i-PrOH), Et3N (1.0 equiv.), reflux for 4 h (for 3b – 50 °C for 2 h). | ||
When unsubstituted benzaldehyde (3a) and 4-chlorobenzaldehyde (3e) were used, it allowed us to obtain a new result of such interactions, namely to get the stable triethylammonium salts 8a and 8e in yields of 35% and 17%, respectively. These salts are uncolored crystalline compounds which can be recrystallized unchanged from ethanol. The possibility of the formation of such salts is caused probably by the raised CH-acidic properties of methyne group (as the result of electron withdrawing influence of SO2-group) which leads to ease of enolization. Moreover, the intramolecular hydrogen bond formation increases the stability of triethylammonium enolates (details of the salt structures are given in X-ray diffraction experimental part). We found only one example in the literature of such three-component reaction wherein similar (but non-symmetrical) salts were isolated.36 These salts contained 4-hydroxycoumarin core and their increased acidic properties was obviously the result of phenolic character of 4-hydroxyl group in addition to the 2-carbonyl group influence.
In the case of benzaldehydes 3c,d, bis-adducts 8c,d were also formed as admixture (12–15% according to 1H NMR) to the target 4H-pyrans 7c,d. Pure products 7c,d were obtained by recrystallization of crude products from ethanol with yields 50 and 39%, correspondingly.
Taking into account the received information, we set the next task to work out an efficient method for the synthesis of 3-ethoxycarbonyl-4H-pyrans 7, which could avoid the formation of undesired bis-adducts 8. Based on the structure of bis-adducts 8, it can be assumed that ethyl cyanoacetate (6) is not involved in the formation of bis-adducts 8 and, consequently, into the three-component interaction, therefore, initially we synthesized arylidene 9 which is presumed to be an intermediate in the synthesis of 2-amino-4H-pyrans37,38 expecting that its reaction with 1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide 1 would allow us to avoid the undesirable formation of bis-adduct 8.
To our surprise, even this stepwise format resulted in the formation of unwanted bis-adduct 8 (usage of arylidene 9a led to 8a with yield of 43%).
To rationalize this result one has to assume that initially formed Michael adduct 10 can be further transformed by two ways (Scheme 2). The first way is the desired heterocyclization, that represents a cascade of proton-transfer processes: enolization of the keto group followed by chain-to-ring tautomeric transformation (hetero-Thorpe–Ziegler reaction, namely intramolecular nucleophilic addition of OH to CN group) and finally enamine–imine tautomerism to form product 7.15 The second one is a retro-Michael reaction with elimination of ethyl cyanoacetate (6) and formation of enone 11, which reacts with the second molecule of benzothiazinone 1 forming bis-adduct 8.
 | ||
| Scheme 2 Presumable mechanism for the formation of compounds 7 and 8. | ||
Enones 11 and arylidenes, similar to them, are the precursors for the synthesis of 2-amino-4H-pyrans reacting with active methylene nitriles.39,40 Arylidenes 11 formed from initial benzothiazinone 1 were described in only one publication41 and obtained under acidic catalysis. Our attempts to obtain arylidenes 11 by interaction of benzothiazinone 1 with benzaldehydes 3a,c (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under the studied reaction conditions (EtOH, 1.0 equiv. of Et3N, rt) led to isolation of bis-adducts 8a,c in low yields and of poor purity. When this reaction was carried out in the molar ratio 2:1 we isolated pure compounds 8a,c in yields of 93% and 87%, correspondingly. This approach can be proposed as preparative synthesis of salts 8. The fact, that interaction 1 with 3 leads to the formation of bis-adducts 8 under conditions of the studied reaction serves as evidence that two pathways of the bis-adducts fromation can accompanay the studied MCR, beside the one discussed above (Scheme 1), an alternative direct route involves a primary formation of enones 11 by interaction of 1 + 3 avoiding the formation of adduct 10. Apparently, a part of the reagents can react by this way.
:1) under the studied reaction conditions (EtOH, 1.0 equiv. of Et3N, rt) led to isolation of bis-adducts 8a,c in low yields and of poor purity. When this reaction was carried out in the molar ratio 2:1 we isolated pure compounds 8a,c in yields of 93% and 87%, correspondingly. This approach can be proposed as preparative synthesis of salts 8. The fact, that interaction 1 with 3 leads to the formation of bis-adducts 8 under conditions of the studied reaction serves as evidence that two pathways of the bis-adducts fromation can accompanay the studied MCR, beside the one discussed above (Scheme 1), an alternative direct route involves a primary formation of enones 11 by interaction of 1 + 3 avoiding the formation of adduct 10. Apparently, a part of the reagents can react by this way.
Thereby, we can conclude that the arylidenes 11 are highly reactive intermediates and behave as Michael acceptors in the reaction media. Depending on the nature of the substituent in the benzaldehyde residue, these arylidenes can interact either with benzothiazinone 1 (to form the symmetrical bis-adduct isolated as triethylammonium salt 8) or with ethyl cyanoacetate 6 (to form desired 4H-pyrans 7).
Since, most probably, all the stages in the mechanism (Scheme 2) are reversible as it follows from the literature data for analogous reactions15,42 (except for 11 → 8, that requires additional investigations), we attempted to take control over the selectivity of this three-component reaction (Scheme 1) by using the excess of ethyl cyanoacetate (6) in our further experiments to shift the reaction equilibrium towards the formation of 4H-pyran derivatives 7.
As noted previously, bis-adduct 8a was the single isolated product when benzaldehyde 3a was used in the three-component interaction (Scheme 1). Using 3.0 equiv. of 6 in the reaction with 1 and 3a in ethanol in the presence of triethylamine in one-pot under reflux during 4 h led to isolation of the desired 4H-pyran derivative 7a. Though the final product was not contaminated with bis-derivative 8a the yields appeared to be moderate (Table 2). Higher amounts of 6 did not affect the yield significantly. These unsatisfactory results compelled us to search for better reaction conditions; we tried to carry out this reaction by varying of the bases applied. In the presence of 1.0 equiv. of 4-dimethylaminopyridine (DMAP) the yield of 7a was increased up to 70%. If lower amounts of DMAP (or Et3N) were used under abovementioned conditions, the reaction was not complete (not shown in the Table 2). The use of DMAP in the case of 4-nitrobenzaldehyde (3b) led to the increase of the yield of 7b up to 68%. For the aldehyde 3c, the pure product 7c was formed in high yield using 2.0 equiv. of 6 (Table 2). The satisfactory yield of 7d was obtained using i-PrOH instead of EtOH. However, for aldehydes 3c,d the use of 1.0 equiv. of DMAP instead of Et3N decreased the reaction efficiency. To avoid the formation of side product 8e and to obtain pure derivative 7e it was necessary to use 7.0 equiv. of 6. In this case, the use of DMAP significantly increased the yield of 7e in comparison with Et3N.
| Aldehyde | Conditions of three-component reaction | Equivalents of 6 | Molar ratio of products 7 and 8b | Yieldsc, % | |
|---|---|---|---|---|---|
| 7 | 8d | ||||
| a The reaction was carried out using 1.0 mmol of 1 and 3 in 5 mL of solvent (EtOH or i-PrOH).b Confirmed by 1H NMR spectra of isolated precipitate without purification as intensity ratio of proton in 4th position of 4H-pyran ring and methyne moiety proton of bis-derivative.c The yields of each component 7 and 8 in the isolated product.d Yields of compound 8 were calculated based on 2.0 equiv. of compound 1.e The admixture of appropriate arylidenes was observed in an amount of 15–17 mol% as was confirmed by 1H NMR spectra of isolated precipitates without purification as intensity ratio of proton in 4th position of 4H-pyran ring and the aldehyde CH. | |||||
| 3a | EtOH, Et3N, reflux for 4 h | 1.0 | 0:1 |
— | 35 |
| 3.0 | 1:0 |
24 | — | ||
| 5.0 | 1:0 |
25 | — | ||
| i-PrOH, Et3N, boiling for 4 h | 1.0 | 1:0.58 |
14 | 16 | |
| 3.0 | 1:0 |
24 | — | ||
| EtOH, morpholine, reflux for 4 h | 1.0 | 1:0.67 |
25 | 34 | |
| 3.0 | 1:0 |
22 | — | ||
| EtOH, DMAP, reflux for 4 h | 3.0 | 1:0 |
70 | — | |
| 3b | EtOH, Et3N, 50 °C, 2 h | 1.0 | 1:0 |
51 | — |
| EtOH, DMAP, 50 °C, 2 h | 1.0 | 1:0 |
68 | — | |
| 3c | EtOH, Et3N, reflux for 4 h | 1.0 | 1:0.16 |
50 | 16 |
| 2.0 | 1:0 |
84 | — | ||
| 3.0 | 1:0 |
78 | — | ||
| EtOH, DMAP, reflux for 4 h | 2.0 | 1:0e |
62 | — | |
| 3d | EtOH, Et3N, reflux for 4 h | 3.0 | 1:0 |
20 | — |
| i-PrOH, Et3N, reflux for 4 h | 1.0 | 1:0.13 |
39 | 10 | |
| 3.0 | 1:0 |
55 | — | ||
| EtOH, DMAP, reflux for 4 h | 3.0 | 1:0e |
62 | — | |
| 3e | EtOH, Et3N, reflux for 4 h | 1.0 | 0:1 |
— | 17 |
| 3.0 | 1:0.36 |
19 | 14 | ||
| 5.0 | 1:0.07 |
29 | 4 | ||
| 7.0 | 1:0 |
33 | — | ||
| EtOH, DMAP, reflux for 4 h | 7.0 | 1:0 |
63 | — | |
Thus, the reaction outcome is strongly dependent on the nature of the substituent in the starting benzaldehyde and the conditions applied. In the case of ethyl cyanoacetate (6) it was necessary to vary the reaction conditions to reach acceptable yield levels.
As it was mentioned above, the mechanism of this reaction (Scheme 2) suggests the formation of the key intermediate 11 that can be transformed into both 4H-pyran derivatives 7 and bis-adducts 8. Though this intermediate was neither isolated, nor identified in the reaction mixtures, its formation was additionally confirmed by studying of mutual transformations of products 5, 7 and 8. Among all the stages of the mechanism, the possibility of reverse transformation of 8 → 11 was unknown. To determine the possibility of the retro-reactions 8 → 7 and 8 → 5, the interactions of bis-adducts 8 with ethyl cyanoacetate (6) and malononitrile (2) were studied. Since the products 8 were isolated for the first time, these experiments allowed us to demonstrate some regularities of their formation and transmutation.
Bis-adducts 8a,c were used to study the transformation of 8 → 7 (Scheme 3). The reactions were carried out by the treatment of 8 with 1.0 equiv. of both ethyl cyanoacetate (6) and triethylamine in refluxing ethanol for 6 h. In the case of 8a (Ar = C6H5) only the starting material was recovered after the heating, whereas when compound 8c (Ar = 2-MeO-C6H4) was used, the final mixture contained the initial bis-adduct 8c, 3-ethoxycarbonyl-4H-pyran 7c and benzothiazinone 1 in molar ratio 1:2:2 (in accordance with 1H NMR spectrum).
To study the transformation of 8 → 5, the bis-adducts 8a,c were used. The reactions of 8a,c with 1.0 equiv. of 2 under the standard reaction conditions resulted in the formation of pure compounds 5a,c isolated in yields 53% and 61% respectively.
Thus, it was experimentally confirmed that the bis-adducts 8 can be transformed into 4H-pyrans 5 and 7 under the studied reaction conditions. This fact clearly indicates the reversibility of the stage 11 ⇄ 8 and consequently all the stages of proposed mechanism (Scheme 2) are reversible.
We have also studied the possibility of reverse transformations of 7 → 8 and 5 → 8. While there is no data about the transformation of such adducts, similar to compounds 8, into 2-amino-4H-pyranes, we found only two suitable references about reverse transformations (similar to 5 or 7 into 8)43,44 which were carried out in AcOH/AcONH4 or under heating in DMF. Transformation of 7 → 8 was achieved in the case of 7a. It was performed by treatment of 7a with 1.0 equiv. of both benzothiazinone 1 and triethylamine in refluxing ethanol for 6 h. As a result, pure bis-adduct 8a was isolated in yield 47%. However, such transformation did not proceed in the case of pyran 7c. The reverse transformation of 5a,c into bis-adducts 8a,c was not achieved under similar conditions (Scheme 3).
 | ||
| Scheme 3 Reagents and conditions: (i) EtOH, Et3N, reflux, 4–6 h; (ii) EtOH, Et3N, 50–60 °C, 1 h. | ||
Taking into account the fact that 3-ethoxycarbonyl-4H-pyran 7a transformed into bis-adduct 8a and 3-cyano-4H-pyran 5a did not, we suggested that malononitrile derived products, 3-cyano-4H-pyrans 5, are more stable compared with products, derived from ethyl cyanoacetate, 3-ethoxycarbonyl-4H-pyrans 7. To confirm this assumption a model reactions 7 → 5 were finally carried out using 7a,c (Scheme 3). Thus, heating of 7a,c at 60 °C for 1 h with 1.0 equiv. of malononitrile (2) in ethanol in the presence of equimolar quantity of triethylamine allowed formation of 5a,c isolated in yield 58% and 67% correspondingly. An example of this type of transformation was previously reported.45 In opposite, the reverse interactions of 5a,c with ethyl cyanoacetate (6) under reflux during 6 h in the presence of equimolar quantity of triethylamine resulted in the recovery of the starting products 5a,c.
Therefore, when ethyl cyanoacetate (6) is used in three-component interaction (Scheme 1) we can conclude that in addition to the reaction conditions, the outcome of the studied MCR is controlled by the relative stability of the products 7 and 8 that is, in its turn, dictated by the substituent nature in the benzaldehyde fragment.
According to our research plan, we also used other active methylene nitriles, such as benzyl cyanide (12) and N-(p-tolyl)cyanoacetamide (13), in this three-component interaction (Scheme 4). But contrary to our expectations, only bis-adducts 8a,b in both cases were isolated in 53% and 44% yields correspondingly. In accordance with the previously established regularities, we used fivefold excess of benzyl cyanide (12) to obtain the target 4H-pyran derivative, but the bis-adduct 8b was again the sole product of this transformation.
 | ||
| Scheme 4 Reagents and conditions: (i) EtOH, Et3N, r.t., 4 h; (ii) EtOH, Et3N, reflux for 4 h. | ||
The IR spectra of all the obtained compounds 5, 7, 8 contain two absorption bands of sulfonyl group at 1329–1296 and 1174–1102 cm−1 regions. The valence oscillations of amino group in compounds 5 and 7 can be observed as two bands at 3456–3285 cm−1. The IR spectra of 5a–g are characterized by the highly intense narrow band of the cyano group which can be found at 2202–2187 cm−1. The frequency of the highly intense band of ester carbonyl function in position 3 of pyran ring is considerably lowered and this band appears in IR spectra of compounds 7 at 1690–1687 cm−1. This is due to the conjugation with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the pyran ring, additionally to the formation of an intramolecular hydrogen bond with the NH2 group.13 The IR spectra of compounds 8 have a series of characteristic absorption bands of the OH-group (broadened band at 3456–3396 cm−1), N+–H-group of triethylammonium cation (2499–2470 cm−1) and bridging CH-group (high intensity band at 3108–2973 cm−1).
C bond of the pyran ring, additionally to the formation of an intramolecular hydrogen bond with the NH2 group.13 The IR spectra of compounds 8 have a series of characteristic absorption bands of the OH-group (broadened band at 3456–3396 cm−1), N+–H-group of triethylammonium cation (2499–2470 cm−1) and bridging CH-group (high intensity band at 3108–2973 cm−1).
The narrow high-intensity singlet of the 4th position of 4H-pyran ring can be observed at 4.48–4.95 ppm in 1H NMR spectra of compounds 5a–f dissolved in DMSO-d6. The same proton signal for compound 5g is observed at 6.54 ppm, that can be explained by unshielding effect of the bulky aromatic anthracene core. The singlet of the 4th position of 4H-pyran ring for 7a–e can be found in the same region, as for compounds 5, 4.78–4.99 ppm. The signal of protons of 2-amino group for pyrans 5a–g is situated in the range of 7.21–7.50 ppm as well as for 7a–e the singlet of the 2-amino group is observed at 7.72–7.94 ppm. The 1H NMR spectra of adducts 8a,b,e are characterized by the presence of singlet of benzothiazine OH-group at 17.10–17.25 ppm and bridging CH-group at 5.69–5.78 ppm. The signals of triethylammonium NH-group are not found in the spectra probably due to the fast deuteroexchange. The 13C NMR spectra of the synthesized compounds reveal the following general features.15 The signal of the C-2 carbon atom of the pyran ring for compounds 5, 7 is found at 158–159 ppm. The peak of the C-3 carbon atom of the pyran ring for compounds 5 is observed at 55–58 ppm as well as for compounds 7 at 58–59 ppm. The C-4 carbon of compounds 5a–g and 7a–e gives signal at 31–37 ppm. The signal at 118–119 ppm was assigned to the nitrile carbon for compounds 5 and the signal at 166–167 ppm was assigned to the carbonyl carbon of the ester group for compounds 7. The 13C NMR spectra of compounds 8 are characterized by the presence of bridged carbon peak at 35–36 ppm.
Molecular ion peaks are observed in mass spectra of 2-amino-4H-pyranes 5 and 7. The mass spectra of bis-adducts 8 are characterized by the presence of the heaviest fragment of ion peak ([M − 326]+) corresponding to enones 11 (Scheme 5).
 | ||
| Scheme 5 Characteristic mass fragmentation of compounds 8. | ||
The structures of compounds 7b and 8b have been additionally confirmed by single crystal X-ray diffraction study (Fig. 3). The compound 7b exists as methanol monosolvate in the crystal phase (more detailed information about molecular and crystal structure see in ESI†). The 8b compound corresponds to the triethylammonium salt with organic anion. The hydrogen atom at the N1s is located from the electron density difference maps. Only one peak corresponding to the hydrogen atom is observed between O1 and O4 atoms from the experimental data and it is located nearer to the O4 atom. The O1–C7 and O4–C11 bond lengths are comparable (1.309(4) Å and 1.314(4) Å, respectively) and slightly shorter than the mean value46 for the Csp2–OH bond (1.333 Å). The O4–H⋯O1 strong charge-assisted hydrogen bond (H⋯O 1.54 Å, O–H⋯O 177°) is formed. Summary, only one tautomer of organic anion with negative charge at the O1 atom may exist in the crystal phase.
 | ||
| Fig. 3 Molecular structure of the compounds 7b and 8b according to the X-ray diffraction study. | ||
3. Conclusion
In this paper the three-component interaction of 1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide with benzaldehydes and active methylene nitriles leading to condensed 2-amino-4-aryl-4H-pyrans was studied. The direction of the three-component reaction is controlled by the nature of both the active methylene nitrile and the substituent in the arylcarbaldehyde: the formation of either 2-amino-4H-pyrans or bis-adducts (or their mixture) was observed. A new product type of such interactions, triethylammonium salts of bis(1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide), was obtained for the first time. The structure and purity of these salts were proved by the single crystal X-ray diffraction study and other instrumental methods. The ability of the bis-adducts to be transformed into 2-amino-4H-pyrans was experimentally confirmed. Based on the known mechanism of the 2-amino-4H-pyrans formation and on the accomplished conversions 7 → 8 and 8 → 7 we proposed the mechanism of the studied MCR which includes a complementary bis-adduct formation stage (Scheme 6). According to the proposed mechanism, arylidenes 11, highly reactive intermediates, that can be transformed further either into the expected 2-amino-4H-pyran derivatives or into the bis-adducts 8, as alternative products (Scheme 6). The reversibility of all stages of the mechanism allowed us to synthesize purposefully the target 2-amino-3-ethoxycarbonyl-4H-pyrans with variable substituent. | ||
| Scheme 6 The general scheme of mutual transformations of compounds 7 and 8. | ||
The found regularities of this three-component reaction can be more general. Currently, there is no other reports regarding the formation of such bis-adducts in similar condensations. Although compounds such as triethylammonium salts of bis(1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide) have not been previously reported in the literature as the possible products of synthesis of 2-amino-4H-pyrans, the potential of their formation should be considered in the study of similar interactions as impurities or as main products.
4. Experimental section
4.1. General
Starting aldehydes and active methylene nitriles were obtained from commercial sources and used without further purification. Starting 1-ethyl-1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide was obtained according to the previously described procedure.5 Arylidenes 4a–g and 9a were prepared via Knoevenagel condensation of the corresponding benzaldehydes 3a–g and active methylene nitriles 2 or 6 in the presence of a base as was reported.47 Dry DMF was prepared in accordance with standard method. Melting points were determined on a Gallenkamp melting point apparatus, model MFB-595 in open capillary tubes. 1H NMR spectra were recorded on Varian Mercury VX 200 instrument or Bruker AMX 500 spectrometer (for 8c) using DMSO-d6 as solvent and TMS as an internal standard. 13C NMR experiments were performed using Bruker AMX 500 spectrometer or Varian Mercury MR-400 (for 7e and 8a). IR spectra were taken on a Perkin-Elmer 298 spectrophotometer in KBr pellets. Elemental analyses were carried out using Carlo Erba CHNS-O EA 1108 analyzer. Mass spectra were taken on a Varian 1200L DIP (EI, 70 eV).4.2. X-ray diffraction experimental part
The crystals of 7b (C22H21N3O7S·CH3OH3) are monoclinic. At 100 K a = 21.5619(7), b = 11.3694(4), c = 18.9777(7) Å, β = 93.367(3)°, V = 4642.4(3) Å3, Mr = 500.77, Z = 1, space group C2/c, dcalc = 1.433 g cm−3, μ(MoKα) = 0.194 mm−1, F(000) = 2096. Intensities of 24414 reflections (6761 independent, Rint = 0.029) were measured on the ![[left double angle bracket]](https://www.rsc.org/images/entities/char_27ea.gif) Xcalibur-3
Xcalibur-3![[right double angle bracket]](https://www.rsc.org/images/entities/char_27eb.gif) diffractometer (graphite monochromated MoKα radiation, CCD detector, ω-scanning, 2Θmax = 60°).
diffractometer (graphite monochromated MoKα radiation, CCD detector, ω-scanning, 2Θmax = 60°).
The crystals of 8b (C6H15NH+·C27H24N3O8S2−) are orthorhombic. At 293 K a = 13.1522(7), b = 23.496(2), c = 11.0242(5) Å, V = 3406.8(3) Å3, Mr = 684.81, Z = 4, space group Pna21, dcalc = 1.335 g cm−3, μ(MoKα) = 0.212 mm−1, F(000) = 1448. Intensities of 33893 reflections (9887 independent, Rint = 0.053) were measured on the Xcalibur-3 diffractometer (graphite monochromated MoKα radiation, CCD detector, ω-scanning, 2Θmax = 60°).
The structures were solved by direct method using SHELXTL package.48 Position of the hydrogen atoms were located from electron density difference maps and refined by “riding” model with Uiso = nUeq of the carrier atom (n = 1.5 for methyl and hydroxyl groups and for water molecule and n = 1.2 for other hydrogen atoms). Full-matrix least-squares refinement of the structures against F2 in anisotropic approximation for non-hydrogen atoms using 6714 (7b), 9840 (8b) reflections was converged to: wR2 = 0.131 (R1 = 0.048 for 5324 reflections with F > 4σ(F), S = 1.046) for structure 7b and wR2 = 0.189 (R1 = 0.067 for 4692 reflections with F > 4σ(F), S = 0.918) for structure 8b. The final atomic coordinates, and crystallographic data for molecules 7b and 8b have been deposited.†
4.3. General procedure for the synthesis of 2-amino-4-aryl-3-cyano-6-ethyl-4,6-dihydropyrano[3,2-c][2,1]benzothiazine 5,5-dioxides (5a–g)
Three-component one-pot procedure. To a solution of 1-ethyl-1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide 1 (0.225 g, 0.001 mol), malononitrile 2 (0.066 g, 0.001 mol) and appropriate benzaldehyde 3a–g (0.001 mol) in ethanol (5–10 mL), triethylamine (0.14 mL, 0.001 mol) was added. The mixture was refluxed during 1 h (in case of 3b the mixture was heated at 50 °C for 40 min). A precipitate usually formed in a few minutes after the beginning of heating. The mixture was cooled to the room temperature; the resulting precipitates of 5a–g were filtered off, washed with ethanol then dried on air and recrystallized from ethanol.
Synthesis using arylidenes 4a–g. To a solution of 1-ethyl-1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide 1 (0.225 g, 0.001 mol) and arylidenes 4a–g (0.001 mol) in ethanol (5–10 mL), triethylamine (0.14 mL, 0.001 mol) was added. The resulted mixture was refluxed for 1 h (in case of 4b mixture was heated at 50 °C for 40 min). During boiling precipitates of 5a–e was gradually formed. The resulting reaction mixtures were treated as mentioned in method A.
The yields for the synthesized compounds 5a–g are presented in Table 1.
Synthesis using 2-amino-3-ethoxycarbonyl-4-R-6-ethyl-4,6-dihydropyrano[3,2-c][2,1]benzothiazine 5,5-dioxides 7a,c. To a solution of 2-amino-3-ethoxycarbonyl-4-R-6-ethyl-4,6-dihydropyrano[3,2-c][2,1]benzothiazine 5,5-dioxides 7a,c (0.001 mol) and malononitrile 2 (0.066 g, 0.001 mol) in ethanol (10 mL), triethylamine (0.14 mL, 0.001 mol) was added. The mixture was heated at 60 °C for 1 h and the resulting solution was cooled to the room temperature. After precipitate of 5a,c was formed it was filtered off, washed with ethanol, dried on air and recrystallized from ethanol. Yields of 5a,c were 58% and 67% correspondingly.
Synthesis using 3-[(4-hydroxy-1-ethyl-2,2-dioxido-1H-2,1-benzothiazin-3-yl)(aryl)methyl]-1-ethyl-1H-2,1-benzothiazin-5-olat 2,2-dioxides 8a,c. To a solution of 3-[(4-hydroxy-1-ethyl-2,2-dioxido-1H-2,1-benzothiazin-3-yl)(aryl)methyl]-1-ethyl-1H-2,1-benzothiazin-5-olat 2,2-dioxides 8a,c and malononitrile 2 (0.066 g, 0.001 mol) in ethanol (10 mL), triethylamine (0.14 mL, 0.001 mol) was added. The mixture was refluxed during 4 h, cooled to 0 °C and kept at this temperature overnight. The resulting precipitate of 5a,c was filtered off, washed with cold ethanol, dried on air and recrystallized from ethanol. Yields of 5a,c were 53% and 61% respectively.
4.4. Procedures for the synthesis of 2-amino-4-aryl-3-ethoxycarbonyl-6-ethyl-4,6-dihydropyrano[3,2-c][2,1]benzothiazine 5,5-dioxides (7a–e)
Compounds 7a–e were obtained by interaction of 1-ethyl-1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide 1, ethyl cyanoacetate 6 and benzaldehydes 3a–e in conditions submitted below. The yields for the synthesized compounds 7a–e are presented in Table 2.White prisms; mp 168–171 °C (from EtOH); anal. calcd for C22H22N2O5S: C, 61.96; H, 5.20; N, 6.57; S, 7.52. Found: C, 62.14; H, 5.08; N, 6.49; S, 7.19; IR (KBr): νmax/cm−1 3419, 3293, 3058, 2982, 2938, 2904, 1951, 1930, 1881, 1812, 1688, 1657, 1617, 1603, 1571, 1520, 1489, 1452, 1320, 1254, 1166, 1147, 1096, 1069, 757 cm−1; 1H NMR (200 MHz, DMSO-d6): δ (ppm) 8.04 (d, J = 7.63, 1H), 7.80 (s, 2H), 7.71–7.49 (m, 2H), 7.39 (t, J = 7.54, 1H), 7.32–7.07 (m, 5H), 4.83 (s, 1H), 4.09–3.79 (m, 4H), 1.11 (t, J = 7.17 Hz, 3H), 0.97 (t, J = 7.23 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ (ppm) 167.2, 159.5, 146.1, 144.4, 137.6, 132.1, 128.2, 127.8, 126.8, 124.5, 123.6, 119.2, 117.2, 116.9, 77.8, 59.2, 42.2, 35.8, 14.3, 13.7; MS (EI) m/z: 426 [M]+.
Yellow prisms; mp 170–172 °C (from EtOH); anal. calcd for C22H21N3O7S: C, 56.04; H, 4.49; N, 8.91; S, 6.80. Found: C, 56.23; H, 4.21; N, 8.60; S, 6.41; IR (KBr): νmax/cm−1 3430, 3396, 3304, 3076, 2976, 2933, 2449, 1949, 1812, 1689, 1657, 1515, 1347, 1316, 1255, 1166, 1146, 1100, 758 cm−1; 1H NMR (200 MHz, DMSO-d6): δ (ppm) 8.18–8.02 (m, 3H), 7.94 (s, 2H), 7.72–7.34 (m, 5H), 4.97 (s, 1H), 4.06–3.79 (m, 4H), 1.10 (t, J = 7.02 Hz, 3H), 0.99 (t, J = 6.87 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ (ppm) 166.9, 159.4, 152.0, 146.5, 146.4, 137.7, 132.5, 129.4, 124.7, 123.7, 123.5, 119.2, 117.0, 115.4, 76.3, 59.4, 42.3, 35.9, 14.2, 13.8; MS (EI) m/z: 471 [M]+.
Colorless prisms; mp 175–177 °C (from EtOH); anal. calcd for C23H24N2O6S: C, 60.51; H, 5.30; N, 6.14; S, 7.02. Found: C, 60.88; H, 5.18; N, 6.35; S, 6.81; IR (KBr): νmax/cm−1 3395, 3287, 3085, 3062, 2983, 2936, 2835, 1959, 1930, 1901, 1805, 1689, 1652, 1615, 1531, 1486, 1463, 1305, 1254, 1163, 1102, 1030, 753 cm−1; 1H NMR (200 MHz, DMSO-d6): δ (ppm) 8.01 (d, J = 6.71 Hz, 1H), 7.72 (s, 2H), 7.67–7.32 (m, 3H), 7.12 (d, J = 7.32 Hz, 2H), 6.94–6.72 (m, 2H), 4.99 (s, 1H), 4.04–3.77 (m, 4H), 3.62 (s, 3H), 1.08 (t, J = 7.17 Hz, 3H), 0.93 (t, J = 6.97 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ (ppm) 167.7, 160.0, 157.6, 146.0, 137.6, 131.8, 131.2, 130.7, 128.3, 124.2, 123.6, 119.9, 119.3, 117.7, 115.6, 111.6, 75.8, 58.9, 55.6, 42.3, 32.4, 14.2, 13.4; MS (EI) m/z: 456 [M]+.
Colorless prisms; mp 175–177 °C (from EtOH); anal. calcd for C23H24N2O6S: C, 60.51; H, 5.30; N, 6.14; S, 7.02. Found: C, 60.85; H, 5.20; N, 6.09; S, 6.77; IR (KBr): νmax/cm−1 3399, 3285, 3072, 2981, 2935, 2901, 2848, 1927, 1844, 1812, 1690, 1656, 1608, 1509, 1450, 1321, 1300, 1255, 1168, 1102, 1032, 756 cm−1; 1H NMR (200 MHz, DMSO-d6): δ (ppm) 8.03 (d, J = 7.94 Hz, 1H), 7.75 (s, 2H), 7.69–7.47 (m, 2H), 7.38 (t, J = 7.53 Hz, 1H), 7.10 (d, J = 8.55 Hz, 2H), 6.80 (d, J = 8.55 Hz, 2H), 4.78 (s, 1H), 4.08–3.81 (m, 4H), 3.67 (s, 3H), 1.13 (t, J = 7.02 Hz, 3H), 1.00 (t, J = 7.17 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ (ppm) 167.3, 159.4, 158.1, 145.8, 137.6, 136.5, 132.1, 128.8, 124.5, 123.6, 119.1, 117.2, 117.1, 113.5, 77.9, 59.2, 55.0, 42.2, 35.0, 14.3, 13.8; MS (EI) m/z: 456 [M]+.
White fine-crystalline powder; mp 198–200 °C (from EtOH); anal. calcd for C22H21ClN2O5S: C, 57.33; H, 4.59; N, 6.08; S, 6.96. Found: C, 57.47; H, 4.33; N, 6.21; S, 6.55; IR (KBr): νmax/cm−1 3432, 3328, 3112, 3047, 2982, 2924, 1687, 1601, 1485, 1458, 1332, 1319, 1253, 1167, 1146, 1118, 780 cm−1; 1H NMR (200 MHz, DMSO-d6): δ (ppm) 8.04 (d, J = 7.93 Hz, 1H), 7.85 (s, 2H), 7.71–7.50 (m, 2H), 7.46–7.16 (m, 5H), 4.83 (s, 1H), 4.07–3.80 (m, 4H), 1.11 (t, J = 7.17 Hz, 3H), 0.99 (t, J = 6.95 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 167.5, 159.7, 146.5, 143.8, 138.0, 132.6, 131.7, 130.1, 128.5, 125.0, 124.0, 119.5, 117.5, 116.7, 77.5, 59.6, 42.6, 35.8, 14.6, 14.1; MS (EI) m/z: 460 [M]+.
4.5. Isolation of 3-[(4-hydroxy-1-ethyl-2,2-dioxido-1H-2,1-benzothiazin-3-yl)(aryl)methyl]-1-ethyl-1H-2,1-benzothiazin-5-olat 2,2-dioxides (8a–c,e)
These products were obtained during our attempts to synthesize the products of three-component interactions using the procedures described below.Procedure A. To a solution of 1-ethyl-1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide 1 (0.225 g, 0.001 mol), ethyl cyanoacetate 6 (0.11 mL, 0.001 mol) and benzaldehyde 3a (0.1 mL, 0.001 mol) in ethanol (5 mL), triethylamine (0.14 mL, 0.001 mol) was added. The mixture was refluxed during 4 h, cooled to the room temperature and solvent was evaporated in vacuum. The oily residue was mixed with 1 mL of i-PrOH. Under intensive mixing the precipitate of 8a was formed, which was filtered off, washed with cold ethanol, dried on air and recrystallized from ethanol. Yield of 8a was 35%.
Procedure B. To a solution of 1-ethyl-1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide 1 (0.225 g, 0.001 mol) and ethyl 2-cyano-3-phenylacrylate 9a (0.201 g, 0.001 mol) in ethanol (5 mL), triethylamine (0.14 mL, 0.001 mol) was added. The mixture was refluxed during 4 h, cooled to 0 °C and kept at this temperature. The precipitate of 8a was formed, washed with cold ethanol, dried on air and recrystallized from ethanol. Yield of 8a was 43%.
Procedure C. Compound 8a was obtained using N-(p-tolyl)cyanoacetamide 13 instead ethyl cyanoacetate 6 under conditions represented in Procedure A. Yield of 8a was 53%.
Procedure D. To a solution of 1-ethyl-1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide 1 (0.225 g, 0.001 mol) and 7a (0.426 g, 0.001 mol) in ethanol (5 mL), triethylamine (0.14 mL, 0.001 mol) was added. The mixture was refluxed during 6 h. Next it was treated as described in Procedure B. Yield of 8a was 47%.
Preparative procedure. To a solution of 1-ethyl-1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide 1 (0.450 g, 0.002 mol), benzaldehyde 3a (0.1 mL, 0.001 mol) in ethanol (10 mL), triethylamine (0.14 mL, 0.001 mol) was added. The solution was stirred at the room temperature. After about 1 h a white precipitate of 8a was formed, this was filtered off, washed with ethanol, dried on air and recrystallized from ethanol. Yield of 8a was 93%.
White needles; mp 153–155 °C (from EtOH); anal. calcd for C33H41N3O6S2: C, 61.95; H, 6.46; N, 6.57; S, 10.02. Found: C, 62.42; H, 6.12; N, 6.76; S, 9.63; IR (KBr): νmax/cm−1 3431, 3087, 2987, 2934, 2882, 2814, 2498, 1613, 1542, 1479, 1310, 1263, 1170, 1141, 1045, 758, 575 cm−1; 1H NMR (200 MHz, DMSO-d6): δ (ppm) 17.25 (s, 1H), 7.86 (d, J = 7.63, 2H), 7.52–7.35 (m, 2H), 7.34–6.97 (m, 9H), 5.71 (s, 1H), 3.95 (q, J = 7.02 Hz, 4H), 3.04 (q, J = 7.32 Hz, 6H), 1.33–1.05 (m, 15H); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 160.4, 141.8, 138.7, 130.5, 128.0, 127.7, 126.8, 125.7, 124.8, 122.1, 117.5, 110.6, 46.2, 41.3, 36.2, 14.5, 9.0; MS (EI) m/z: 313 [M − 326]+.
Yellow prisms; mp 168–170 °C (from EtOH); anal. calcd for C33H40N4O8S2: C, 57.88; H, 5.89; N, 8.18; S, 9.36. Found: C, 58.12; H, 5.94; N, 8.40; S, 9.14; IR (KBr): νmax/cm−1 3396, 3108, 2985, 2935, 2496, 1606, 1514, 1480, 1347, 1308, 1254, 1163, 1143, 1109, 1047, 751, 567 cm−1; 1H NMR (200 MHz, DMSO-d6): δ (ppm) 17.10 (s, 1H), 8.09 (d, J = 8.85, 2H), 7.86 (d, J = 6.41 Hz, 2H), 7.54–7.39 (m, 4H), 7.36–7.27 (m, 2H), 7.20–7.07 (m, 2H), 5.78 (s, 1H), 3.96 (q, J = 6.71 Hz, 4H), 3.05 (q, J = 7.32 Hz, 6H), 1.29–1.05 (m, 15H); 13C NMR (125 MHz, DMSO-d6): δ (ppm) 160.4, 150.3, 145.7, 138.4, 130.5, 128.5, 126.5, 124.2, 123.1, 121.9, 117.4, 109.5, 45.9, 41.1, 36.3, 14.1, 8.7; MS (EI) m/z: 358 [M − 326]+.
Preparative procedure. To a solution of 1-ethyl-1H-2,1-benzothiazin-4(3H)-one 2,2-dioxide 1 (0.450 g, 0.002 mol), 2-methoxybenzaldehyde 3c (0.136 g, 0.001 mol) in ethanol (10 mL), triethylamine (0.14 mL, 0.001 mol) was added. The solution was stirred at the room temperature. After about 1 h a white precipitate of 8c was formed, this was filtered off, washed with ethanol, dried on air and recrystallized from ethanol. Yield of 8c was 87%.
Colorless prisms; mp 145–147 °C (from EtOH); anal. calcd for C34H43N3O7S2: C, 60.96; H, 6.47; N, 6.27; S, 9.57. Found: C, 61.31; H, 6.46; N, 6.24; S, 9.22; IR (KBr): νmax/cm−1 3429, 2973, 2917, 2850, 2470, 1606, 1587, 1474, 1335, 1296, 1247, 1164, 1143, 1108, 1046, 744, 567 cm−1; 1H NMR (500 MHz, DMSO-d6): δ (ppm) 16.84 (s, 1H), 7.99–7.84 (m, 2H), 7.52–7.35 (m, 3H), 7.33–7.00 (m, 5H), 6.84–6.70 (m, 2H), 5.89 (s, 1H), 4.06–3.81 (m, 4H), 3.62 (s, 3H), 3.11–2.90 (m, 6H), 1.39–0.96 (m, 15H); 13C NMR (125 MHz, DMSO-d6): δ (ppm) 160.0, 156.6, 138.9, 130.4, 129.9, 129.8, 126.8, 126.4, 124.9, 121.6, 119.0, 117.5, 110.6, 110.4, 55.2, 45.9, 41.8, 31.9, 14.3, 8.7.
Colorless prisms; mp 163–165 °C (from EtOH); anal. calcd for C33H40ClN3O6S2: C, 58.78; H, 5.98; N, 6.23; S, 9.51. Found: C, 58.91; H, 5.87; N, 6.41; S, 9.34; IR (KBr): νmax/cm−1 3456, 3079, 2988, 2937, 2904, 2878, 2835, 2499, 1610, 1542, 1486, 1311, 1263, 1170, 1139, 1113, 1047, 757, 571 cm−1; 1H NMR (200 MHz, DMSO-d6): δ (ppm) 17.19 (s, 1H), 7.86 (d, J = 7.94, 2H), 7.51–7.36 (m, 2H), 7.34–7.19 (m, 6H), 7.18–7.06 (m, 2H), 5.69 (s, 1H), 3.95 (q, J = 7.22 Hz, 4H), 3.03 (q, J = 7.32 Hz, 6H), 1.28–1.05 (m, 15H); 13C NMR (125 MHz, DMSO-d6): δ (ppm) 160.1, 140.6, 138.4, 130.3, 130.0, 129.2, 127.6, 126.5, 124.3, 121.8, 117.2, 110.0, 45.9, 40.9, 35.5, 14.1, 8.7; MS (EI) m/z: 347 [M − 326]+.
Acknowledgements
Authors are thankful to Mr Maxim A. Nechayev (Enamine Ltd) and Ms Maria A. Vodolazhenko (SSI “ISC” NASU) for measurement of 13C NMR spectra. We also acknowledge Mr Aleksey L. Shemchuk and Mr Pavel S. Arzumanov for their valuable remarks.References
- H. Bienaymé, C. Hulme, G. Oddon and P. Schmitt, Chem.–Eur. J., 2000, 6, 3321–3329 CrossRef.
- B. Jiang, S.-J. Tu, P. Kaur, W. Wever and G. Li, J. Am. Chem. Soc., 2009, 131, 11660–11661 CrossRef CAS PubMed.
- C. Hulme and V. Gore, Curr. Med. Chem., 2003, 10, 51–80 CrossRef CAS PubMed.
- (a) Y. V. Sedash, N. Y. Gorobets, V. A. Chebanov, I. S. Konovalova, O. V. Shishkin and S. M. Desenko, RSC Adv., 2012, 2, 6719–6728 RSC; (b) V. A. Chebanov and S. M. Desenko, Chem. Heterocycl. Compd., 2012, 48, 566–583 CrossRef CAS; (c) V. A. Chebanov, V. E. Saraev, S. M. Desenko, V. N. Chernenko, S. V. Shishkina, O. V. Shishkin, K. M. Kobzar and C. O. Kappe, Org. Lett., 2007, 9, 1691–1694 CrossRef CAS PubMed; (d) N. Y. Gorobets, Y. V. Sedash, K. S. Ostras, O. V. Zaremba, S. V. Shishkina, V. N. Baumer, O. V. Shishkin, S. M. Kovalenko, S. M. Desenko and E. V. Van der Eycken, Tetrahedron Lett., 2010, 51, 2095–2098 CrossRef CAS; (e) E. A. Muravyova, S. M. Desenko, R. V. Rudenko, S. V. Shishkina, O. V. Shishkin, Y. V. Sen'ko, E. V. Vashchenko and V. A. Chebanov, Tetrahedron, 2011, 67, 9389–9400 CrossRef CAS.
- L. A. Shemchuk, D. A. Lega, R. G. Redkin, V. P. Chernykh, O. V. Shishkin and S. V. Shishkina, Tetrahedron, 2014, 70, 8348–8353 CrossRef CAS.
- J. G. Lombardino and N. W. Treadway Jr, Org. Prep. Proced. Int., 1971, 3, 33–36 CrossRef CAS.
- F. T. Coppo and M. M. Fawzi, J. Heterocycl. Chem., 1998, 35, 983–987 CrossRef CAS.
- M. Shafiq, M. Zia-Ur-Rehman, I. U. Khan, M. N. Arshad and S. A. Khan, J. Chil. Chem. Soc., 2011, 56, 527–531 CrossRef CAS.
- Y. Misu and H. Togo, Org. Biomol. Chem., 2003, 1, 1342–1346 CAS.
- I. V. Ukrainets, L. A. Petrushova and S. P. Dzyubenko, Chem. Heterocycl. Compd., 2013, 49, 1378–1383 CrossRef CAS.
- I. V. Ukrainets, L. A. Petrushova, S. P. Dzyubenko and Y. Liu, Chem. Heterocycl. Compd., 2014, 50, 564–572 CrossRef CAS.
- I. V. Ukrainets, L. A. Petrushova, S. P. Dzyubenko and G. Sim, Chem. Heterocycl. Compd., 2014, 50, 103–110 CrossRef CAS.
- G. Brahmachari and B. Banerjee, ACS Sustainable Chem. Eng., 2013, 2, 411–422 CrossRef.
- H. R. Shaterian, M. Arman and F. Rigi, J. Mol. Liq., 2011, 158, 145–150 CrossRef CAS.
- Y. M. Litvinov and A. M. Shestopalov, in Adv. Heterocycl. Chem., ed. A. R. Katritzky, Academic Press, 2011, vol. 103, pp. 175–260 Search PubMed.
- R. Ghahremanzadeh, G. Hosseini, R. Akbarzadeh and A. Bazgir, J. Heterocycl. Chem., 2013, 50, 272–280 CrossRef CAS.
- A. A. Shestopalov, L. A. Rodinovskaya, A. M. Shestopalov and V. P. Litvinov, Russ. Chem. Bull., 2004, 53, 724–725 CrossRef CAS.
- D. Heber and E. V. Stoyanov, Synthesis, 2003, 2003, 0227–0232 CrossRef.
- D.-O. Moon, Y. H. Choi, N.-D. Kim, Y.-M. Park and G.-Y. Kim, Int. Immunopharmacol., 2007, 7, 506–514 CrossRef CAS PubMed.
- Z.-Q. Xu, M. G. Hollingshead, S. Borgel, C. Elder, A. Khilevich and M. T. Flavin, Bioorg. Med. Chem. Lett., 1999, 9, 133–138 CrossRef CAS PubMed.
- M. Makino and Y. Fujimoto, Phytochemistry, 1999, 50, 273–277 CrossRef CAS.
- S. Ray, H. K. Majumder, A. K. Chakravarty, S. Mukhopadhyay, R. R. Gil and G. A. Cordell, J. Nat. Prod., 1996, 59, 27–29 CrossRef CAS PubMed.
- Y.-L. Lin, C.-C. Shen, Y.-J. Huang and Y.-Y. Chang, J. Nat. Prod., 2005, 68, 381–384 CrossRef CAS PubMed.
- E. E. Schweizer and D. Meeder-Nycz, in Chem. Heterocycl. Comp., John Wiley & Sons, Inc., 2008, pp. 11–139 Search PubMed.
- D. Armesto, W. M. Horspool, N. Martin, A. Ramos and C. Seoane, J. Org. Chem., 1989, 54, 3069–3072 CrossRef CAS.
- B. Pirotte, J. Fontaine and P. Lebrun, Curr. Med. Chem., 1995, 2, 573 CAS.
- K. S. Atwal, Curr. Med. Chem., 1996, 3, 227 CAS.
- C. S. Konkoy, D. B. Fick, S. X. Cai, N. C. Lan and J. F. W. Keana, US Pat., 6 680 332 B1, 2004.
- S. Kang, G. Cooper, S. F. Dunne, C.-H. Luan, D. James Surmeier and R. B. Silverman, Bioorg. Med. Chem., 2013, 21, 4365–4373 CrossRef CAS PubMed.
- A.-G. E. Amr, A. M. Mohamed, S. F. Mohamed, N. A. Abdel-Hafez and A. E.-F. G. Hammam, Bioorg. Med. Chem., 2006, 14, 5481–5488 CrossRef CAS PubMed.
- P. Paliwal, S. Jetti and S. Jain, Med. Chem. Res., 2013, 22, 2984–2990 CrossRef CAS.
- D. Kumar, V. B. Reddy, S. Sharad, U. Dube and S. Kapur, Eur. J. Med. Chem., 2009, 44, 3805–3809 CrossRef CAS PubMed.
- C. W. Smith, J. M. Bailey, M. E. J. Billingham, S. Chandrasekhar, C. P. Dell, A. K. Harvey, C. A. Hicks, A. E. Kingston and G. N. Wishart, Bioorg. Med. Chem. Lett., 1995, 5, 2783–2788 CrossRef CAS.
- T. A. Nakib, V. Bezjak, S. Rashid, B. Fullam and M. J. Meegan, Eur. J. Med. Chem., 1991, 26, 221–230 CrossRef CAS.
- Y. A. Sharanin, L. Y. Sukharevskaya and V. V. Shelyakin, Russ. J. Org. Chem., 1998, 34, 552–553 CAS.
- M. P. Goncharenko and Y. A. Sharanin, Russ. J. Org. Chem., 1993, 29, 1218–1229 Search PubMed.
- Z. H. Khalil, A. A. Abdel-Hafez, A. A. Geies and A. M. Kamal El-Dean, Bull. Chem. Soc. Jpn., 1991, 64, 668–670 CrossRef CAS.
- L. Rodinovskaya, A. Shestopalov, A. Gromova and A. Shestopalov, Synthesis, 2006, 2006, 2357–2370 CrossRef.
- N. Martín, A. Martínez-Grau, C. Seoane and J. Marco, Tetrahedron: Asymmetry, 1995, 6, 255–262 CrossRef.
- A. M. Shestopalov and O. A. Naumov, Russ. Chem. Bull., 2003, 52, 961–968 CrossRef CAS.
- J. L. Hicks and W. H. Roark, Pat. WO 2004/014388 A1, 2004.
- A. M. Shestopalov and Y. M. Emel'yanova, in Selected methods for synthesis and modification of heterocycles (Russ. Transl.), ed. V. G. Kartsev, IBS Press, 2003, vol. 2, pp. 534–563 Search PubMed.
- H. A. A. El-Nabi, Pharmazie, 1997, 52, 28–32 Search PubMed.
- R. A. Mekheimer, N. H. Mohamed and K. U. Sadek, Bull. Chem. Soc. Jpn., 1997, 70, 1625–1630 CrossRef CAS.
- M. R. H. Elmoghayer, M. A. E. Khalifa, M. K. A. Ibraheim and M. H. Elnagdi, Monatsh. Chem., 1982, 113, 53–57 CrossRef CAS.
- Structure correlation, ed. H.-B. Burgi and J. D. Dunitz, VCH, Weinheim, 1994, vol. 2, pp. 741–784 Search PubMed.
- R. G. Redkin, L. A. Shemchuk, V. P. Chernykh, O. V. Shishkin and S. V. Shishkina, Tetrahedron, 2007, 63, 11444–11450 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full spectroscopic data (1H and 13C NMR, IR, MS) for compounds 5, 7 and 8 and X-ray crystallographic data for compounds 7b and 8b. CCDC 1405511 and 1405512. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra24566d |
| This journal is © The Royal Society of Chemistry 2016 |