
Spectroscopic tracking of mechanochemical reactivity and modification of a hydrothermal char†
Abstract
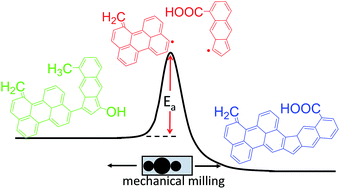
A glucose hydrothermal char (HTC) was synthesized and ball milled to break chemical bonds, generate defects, and form new chemical structures. To follow the chemical transformations, the physical, chemical, and structural characteristics of the milled HTC were studied using a variety of imaging and spectroscopic techniques. Scanning electron microscopy showed that milling initially induced agglomeration of HTC microspheres accompanied by a more gradual shift in particle shape from spherical to plate-like. The elemental composition of the synthesized HTC was comparable to lignite; ball milling converted the material to a more coal-like composition. Infra-red spectroscopy indicated increasing aromatic and carboxylic acid content after milling. Raman microspectroscopy indicated an increase in graphitization during ball milling and a decrease in edge sites. ESR measurements indicated the production of carbon radicals with ball milling that reached a maximum after 90 min. Based on these results, we outline a mechanism of mechanochemical HTC modification whereby weak aliphatic and ether bonds are cleaved to form edge sites bearing carbon radicals, which then recombine to form graphene basal planes. In the transition state, the surface composition of hydrothermal char is readily modified – here, we demonstrate introduction of carboxylic acids as surface oxygen groups.
 Please wait while we load your content...
Please wait while we load your content...

