
Improved electrochemical properties of tavorite LiFeSO4F by surface coating with hydrophilic poly-dopamine via a self-polymerization process†
Abstract
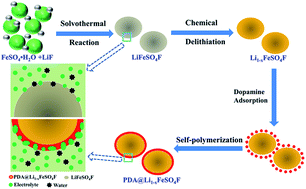
Poly-dopamine coated Li1−xFeSO4F is prepared via a self-polymerization process. The material shows larger discharge capacities, better rate capability and longer cycle stability than the pristine LiFeSO4F. The improved electrochemical properties are attributed to the highly hydrophilic and elastic properties of poly-dopamine.
 Please wait while we load your content...
Please wait while we load your content...

