
Theranostic liposomes containing conjugated polymer dots and doxorubicin for bio-imaging and targeted therapeutic delivery†
Man Ma,
Mingzhu Lei,
Xiaoxiao Tan,
Fengping Tan* and
Nan Li*
Tianjin Key Laboratory of Drug Delivery & High-Efficiency, School of Pharmaceutical Science and Technology, Tianjin University, 300072, Tianjin, P. R. China. E-mail: linan19850115@163.com; tanfengping@163.com; Tel: +86-22-27404986 Tel: +86-22-27405160
First published on 21st December 2015
Abstract
This work was devoted to the development of a lipid-based theranostic nanoparticle able to simultaneously host conjugated polymer dots, doxorubicin (Dox) and folate acid (FA). Poly(9,9-dioctylfluorene-2,7-diyl-co-benzothiadiazole) (PFBT) was chosen as the fluorescent probe because of its high brightness in in vitro cellular uptake studies and good biocompatibility in in vitro/in vivo toxicity experiments. The theranostic liposomes (PFBT–Dox–Lip–FA) exhibited a hydrodynamic size of 127.30 ± 3.20 (nm) with a zeta potential of −25.00 ± 2.00 (mV). Mostly importantly, the extent of Dox release at 24 h from PFBT–Dox–Lip–FA showed a satisfactory result under mild hyperthermia conditions compared with Dox–Lip–FA. Such rapid release led to a lower half maximal inhibitory concentration (IC50) in MCF-7 cells at 16.8 ± 4.5 (μg mL−1), whereas the IC50 of Dox–Lip–FA (37 °C) was 28.3 ± 3.7 (μg mL−1). The cellular uptake study also revealed higher drug accumulation in tumor cells for theranostic liposomes. In vivo studies of PFBT–Dox–Lip–FA on tumor-bearing mouse models revealed that the distribution of liposomes in the tumors could be indicated accurately by PFBT. Besides, tumor-bearing mice could be significantly inhibited by PFBT–Dox–Lip–FA. Together with its negligible in vivo toxicity, PFBT–Dox–Lip–FA is a useful system for simultaneous cancer diagnosis and targeted drug delivery.
Introduction
Cancer has been one of the major threats to the lives of human beings for centuries.1,2 The informative and accurate visualization of anti-cancer agents, following their administration, can play a considerable and indispensable role to formulate effective anti-cancer therapeutics.3 Conventional diagnostic probes and small molecular therapeutic agents are often limited by undesirable differences in biodistribution and selectivity. Therefore, many efforts have been performed in the areas of multifunctional theranostic nanoparticles, which integrated targeting, therapeutic, and diagnostic functions.4,49Theranosis is a newly emerging concept which involves simultaneous execution of diagnostic tests and targeted therapy.4,48 It requires noninvasive imaging modalities including fluorescence imaging,5 magnetic resonance imaging,6 and nuclear imaging.7 In particular, fluorescence imaging has attracted considerable research interest as the technique is safe, inexpensive and time-dependent with excellent temporal resolution,8 which also requires less resources and space than nuclear imaging and magnetic resonance imaging.9 To date, various fluorescent materials including small organic dyes,10 fluorescent proteins,11 semiconductor quantum dots (QDs)12 as well as carbon nanotubes,13 have been employed as fluoprobes for in vivo fluorescence imaging. However, organic dyes and proteins have a great number of drawbacks, such as low molar absorptivity and poor photostability. As compared to fluorescent proteins and organic dyes, QDs have shown higher brightness and better photostability in vitro as well as in vivo studies.14 But for in vivo imaging, the intrinsically toxic components in QDs (e.g., cadmium and selenium) that can be released in biological media may cause serious damage to living bio-substrates.15 As a result, exploration of novel fluorescent probes with high brightness, good photostability and low systemic toxicity is highly desirable for in vivo applications.
In this regard, conjugated polymers, a class of fluorescent macromolecules with highly delocalized π-conjugated backbones, have attracted great research interests owing to their unique photochemical and electroluminescence characteristics, such as light harvesting properties, large extinction coefficient, high brightness, and good biocompatibility.16 Recently, great research interest has also been focused on the application of conjugated polymers for in vivo bio-imaging.17 To cite just a representative example, a recent investigation has demonstrated that conjugated polymers-based theranostic nanoparticles could be of great help for in vivo imaging through intravenous injection.18 As most organic-soluble conjugated polymers are highly hydrophobic, the main obstacle for conjugated polymers in bio-imaging applications is how to enhance their dispersibility in water. Based on the prior studies, this can be achieved through either chemical modification to produce water-soluble conjugated polyelectrolytes19 or simply by physical approach via mini-emulsion, nano-precipitation, solvent extraction single emulsion, encapsulation, etc. to fabricate water-dispersible nanoparticles.20–22 As compared to the synthesis of conjugated polyelectrolytes, fabrication of conjugated polymers in nanoparticles serves as a much simple and general strategy seems to be a much simpler strategy that can potentially be applied more commonly, as well.
So far, various nanoparticle-based carriers have been proposed for theranostic applications including liposomes,23 polymeric micelles and nanogels.24 Among them, liposomes have attracted considerable interest because of their biocompatibility, biodegradability, low toxicity, and aptitude to trap both hydrophilic and lipophilic drugs.25 What's more, they could simplify site-specific drug delivery to tumor tissues.26
In this contribution, we reported a conjugated polymer dots embedded and Dox loaded lipid theranostic nanoparticle (PFBT–Dox–Lip–FA), which allowed for simultaneous in vivo bio-imaging and targeting drug delivery in a tumor-bearing mouse model. PFBT was selected because it had an absorption maximum at 440 nm, matching the 457 nm laser equipped on the confocal microscopy. In addition, it showed a large Stokes shift of 117 nm, which was ideal for fluorescence imaging.18 Small molecule Dox was one of the most important anticancer agents for solid tumor.27 FA has emerged as an optimal targeting ligand for selective delivery of attached imaging and therapeutic agents to cancer tissues and sites of inflammation.28 In this study, the embedding of hydrophobic PFBT into the bilayers enhanced its solubility and biocompatibility, further realized the efficiently the embedding of PFBT into the bilayers rendered hydrophobic PFBT biocompatible and efficiently labeled of tumor cells in vitro and in vivo. In the present work, we offered further systematic characterization and in vitro/in vivo evaluation of the PFBT–Dox–Lip–FA hybrid vesicles. In this way, the potential use of PFBT–Dox–Lip–FA hybrids was explored as theranostic devices for the simultaneous targeting delivery of therapeutic and diagnostic agents.
Experimental section
Materials
Doxorubicin hydrochloride (Dox, MW 579.98, 99% purity) was obtained from Fisher Scientific (Waltham, MA, USA). 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-distearoylsn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (DSPE-PEG2000) and DSPE-PEG2000–FA were purchased from Ruixi Biological Technology Co., Ltd (Xi'an, China). Poly(9,9-dioctylfluorene-2,7-diyl-co-benzothiadiazole) (PFBT) was kindly provided by Dr D. Ding from State Key Laboratory of Medicinal Chemical Biology, Nankai University. Cholesterol, 3,3′-dioctadecyloxacarbocyanine perchlorate (Dio), 4′,6-diamidino-2-pheylindole (DAPI) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma (St Louis, MO). All other chemicals and reagents used were of analytical grade.Liposome preparation
Liposomes were prepared by the film hydration method and Dox was encapsulated into the liposome by the ammonium sulfate gradient method.29 Briefly, DPPC, DSPE-PEG2000–FA and cholesterol in chloroform were mixed in a round-bottom flask in a ratio of DPPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cholesterol:DSPE-PEG2000–FA = 67:30:3. The total mass of lipids used was 30 mg. Then the mixture was evaporated to remove the solvent and fabricate a thin film. The obtained thin film was dispersed in 300 mM ammonium sulfate and hydrated by gentle vortexing. The suspended solution was extruded in a series with 800 nm, 400 nm, 200 nm, and 100 nm pore-sized membranes. The free ammonium sulfate was removed by dialysis for 6 h at room temperature. Then Dox, at a drug-to-lipid molar ratio of 1:15, was dissolved in the solution and incubated at 37 °C for 6 h. The Dox-loaded liposomes were stored at 4 °C for later use. Unentrapped drug was removed by dialysis against 0.9% NaCl. After separation, the amount of Dox entrapped within the liposomes was determined by fluorescence spectrometer method (Ex = 480 nm, Em = 560 nm). The standard curve of the absorbance of Dox at 480 nm versus the Dox concentration was plotted with a very good linear correlation: Y = 11.63X + 43.70 (1.0–20.0 μg mL−1), r2 = 0.9993, with the recovery of 99.5%. The encapsulation efficiency (EE, %) of Dox was estimated from the following formula:
:cholesterol:DSPE-PEG2000–FA = 67:30:3. The total mass of lipids used was 30 mg. Then the mixture was evaporated to remove the solvent and fabricate a thin film. The obtained thin film was dispersed in 300 mM ammonium sulfate and hydrated by gentle vortexing. The suspended solution was extruded in a series with 800 nm, 400 nm, 200 nm, and 100 nm pore-sized membranes. The free ammonium sulfate was removed by dialysis for 6 h at room temperature. Then Dox, at a drug-to-lipid molar ratio of 1:15, was dissolved in the solution and incubated at 37 °C for 6 h. The Dox-loaded liposomes were stored at 4 °C for later use. Unentrapped drug was removed by dialysis against 0.9% NaCl. After separation, the amount of Dox entrapped within the liposomes was determined by fluorescence spectrometer method (Ex = 480 nm, Em = 560 nm). The standard curve of the absorbance of Dox at 480 nm versus the Dox concentration was plotted with a very good linear correlation: Y = 11.63X + 43.70 (1.0–20.0 μg mL−1), r2 = 0.9993, with the recovery of 99.5%. The encapsulation efficiency (EE, %) of Dox was estimated from the following formula:
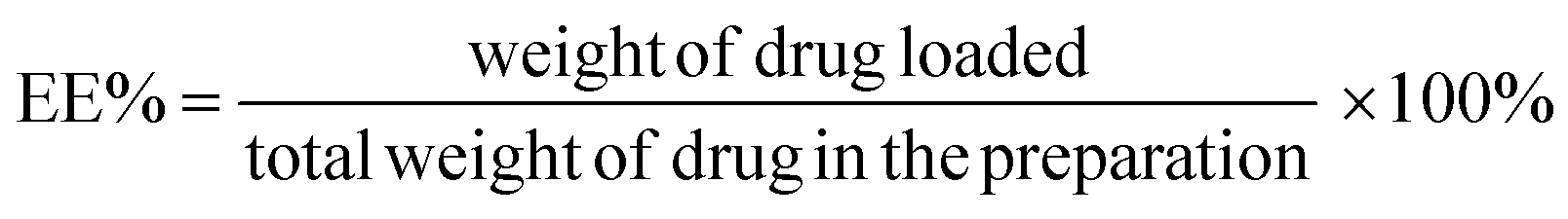 | (1) |
The experiments were conducted in triplicate and data were expressed as means ± SD.
For the preparation of fluoprobe-loaded liposomes, the above lipids and fluoprobe (PFBT or Dio) were dissolved in chloroform and evaporated under reduce pressure. The obtained thin film was hydrated using the same method as described before. The fluoprobe-loaded liposomes were stored at 4 °C for later use.
Characterization of test liposomes
The particle size, polydispersity index (PDI), and zeta-potential of the test liposomes were determined by the dynamic light scattering (DLS) (Malvern Instruments, Malvern, UK). The morphology of vesicles was observed with a JEM-100 CX transmission electron microscope (Jeol Ltd., Tokyo, Japan) followed by negative staining procedure using phosphotungstic acid. Atomic force microscopy (AFM, Nanowizarc, JPK Ltd., Berlin, Germany) was used to further observe the liposomal surface morphology. The formulation was diluted with distilled water and was then spread on a mica sheet, dried at room temperature. UV-vis spectra was collected on Shimadzu UV-1700 spectrometer. Fluorescence microscopic images were acquired by Leica TCS SP5 Inverted Supercontinuum Confocal Laser Scanning Microscope (CLSM) (Leica Microsystems, Heidelberg, Germany).In vitro release studies
Temperature-dependent release profile of Dox from Dox–Lip–FA was established by fluorometry upon incubating the samples at various temperatures from 37 °C to 42 °C for 30 min. Typically, 1 mL Dox–Lip–FA was dialyzed (Mw cutoff 12000–14000 Da) against 50 mL of release media (phosphate buffer, pH 7.4). At selected time intervals, 1 mL release media was taken out and replenished with an equal volume of fresh media. The amount of released Dox was determined by fluorometry as described before. Dox release rate (%) was calculated as follows:
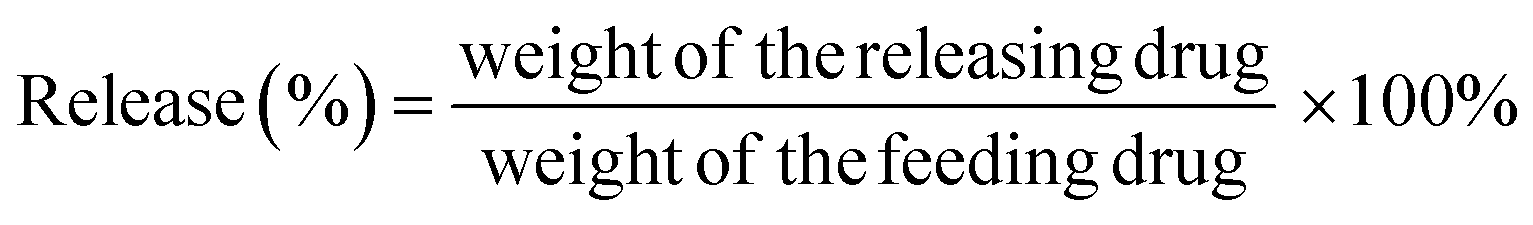 | (2) |
Release of Dox from PFBT–Dox–Lip–FA as a function of time was performed at 37 °C and 42 °C, respectively. Dox–Lip–FA samples were considered as a control. The Dox release (%) was also calculated as before.
Cell culture
In this work, HepG2 cells and MCF-7 cells were used to evaluate the targeting effect of the constructed nano-carrier, as the two cell lines were reported to be of lack FR expression and FR overexpression, respectively. They were received as a gift from the institute of radiation medicine Chinese academy of medical sciences (Tianjin, China). The cells were cultured in Dulbecco's modified essential medium (DMEM) with 1% L-glutamine, 10% FBS, 100 IU mL−1 penicillin, and 100 IU mL−1 streptomycin (all Gibco).47 Cultures were maintained in a humidified atmosphere at 37 °C in 5% CO2. Growth media was replaced every three days and cellular confluence and morphology were periodically monitored.In vitro cytotoxicity assay
Cytotoxicity of fluoprobe loaded liposomal formulations was tested against both HepG2 and MCF-7 cells. Briefly, each well of 96-well plates was seed with 5 × 103 cells and incubated for 24 h. Then the cells were exposed to two samples (PFBT–Lip–FA or Dio–Lip–FA) at different concentrations (0.05–5 μM) for 24 h at 37 °C. Both liposomal samples were transferred by the culture medium. Cytotoxicity of Dox loaded liposomal formulations at different temperature was also performed with the two cell lines. After respectively adding free Dox, Dox–Lip–FA or PFBT–Dox–Lip–FA to the cells, the 96-well plates were immediately placed in a water bath at mild hyperthermia (42 °C) (plates were vacuum sealed to be waterproof) or in an incubator at 37 °C for 1 h. Then the cells were incubated in the medium at 37 °C for 24 h. Finally, 20 μL of MTT solution (5 mg mL−1) was added to each well, followed by incubation for another 4 h at 37 °C. The precipitated formazan was dissolved in 200 μL of DMSO. The absorbance was read on power wave × 340 Microplate Reader (Bio-tek instruments Inc., USA) at wavelength of 570 nm. The data reported represented the means of triplicate measurement. The percentage of cell viability was determined by comparing cells treated with drug with the untreated control cells. The IC50 was calculated using the Origin software package (OriginLab, Northsampton, MA, USA).In vitro cellular uptake
A confocal fluorescent microscope was used to compare the intracellular distribution and cellular uptake of different liposomes. To observe the cellular delivery of Dox-loaded liposomes with or without FA-targeting delivery system, both HepG2 and MCF-7 cells were seeded in the 35 mm cell culture plates at a density of 1 × 104 cells per well in 1 mL culturing medium at 37 °C for 24 h till 80% confluent, respectively.Then, the medium was removed and replaced with 1 mL dual-loaded formulations with Dox/fluoprobe (PFBT or Dio) concentration of 10 μg mL−1 and 10 μM, respectively. After a predetermined time course (1 h or 4 h), the extra medium was discarded. The cells were washed with serum free media (1 mL) in triplicate, and then fixed with 4% paraformaldehyde for 20 min, followed by cell nuclei staining with DAPI (1 μg mL−1) for 20 min. After that, the DAPI was carefully cleared with PBS for three times. By adding 1 mL PBS, the cells were imaged by CLSM. The fluorescence intensity of the obtained images was quantified by Image Pro-Plus Software 6.0 (Media Cybernetics, USA).
For the temperature sensitivity assay, medium was then refreshed with PFBT–Dox–Lip–FA suspension in serum-free medium. The cells were incubated at 37 °C (control) or 42 °C (mild hyperthermia) for 1 h (it took 10 min to reach medium temperature of 42 °C). The CLSM images were obtained as described before.
Tumor models
Female nude mice (18–20 g) were purchased from Beijing Vital River Company (Beijing, China) and housed under standard conditions with free access to food and water. The animals were conducted in accordance with the Animal Management Rules of the Ministry of Health of the People's Republic of China (Document no. 55, 2001) and protocols approved by the School of Pharmaceutical Science and Technology, Tianjin University. The tumor-bearing nude mice models were prepared by subcutaneously injecting 6 × 106 MCF-7 cells/100 μL PBS into the right oxter of the mice.In vivo biodistribution study
When the tumors were approximately 200 mm3, the mice were randomly divided into 5 groups (three per group) and intravenously injected with Dio–Lip–FA, PFBT–Lip–FA, Dox–Lip–FA, and PFBT–Dox–Lip–FA, respectively (the temperature was 37 °C or 42 °C, and the Dox/fluoprobe dose was 5 mg kg−1 and 1 mg kg−1). For the temperature sensitivity study, the mice were anesthetized, and the right legs were immersed into a water bath maintained at 42 °C for 10 min to equilibrate the temperature before the injection of PFBT–Dox–Lip–FA. The temperature inside the tumor (40–42 °C) was verified by a thermocouple (HH306 data logger/thermometer, Omega engineering, Inc. Stamford, CT). The hyperthermia was maintained for another 1 h after the injection. In contrast, the group of PFBT–Dox–Lip–FA at 37 °C was carried out without heat. Images were taken on IVIS Lumina imaging system (Caliper, USA) at 2, 6 and 24 h post injection. The light with a central wavelength at 455 nm was selected as the excitation source. In vivo spectral imaging from 500 nm to 900 nm (10 nm step) was carried out with an exposure time of 1000 ms for each image frame. After the 24 h imaging, mice were euthanized and tissue samples (liver, spleen, lung, kidney, heart and tumor) were collected for ex vivo imaging. The fluorescence intensities were analyzed by Living Image Software.In vivo antitumor efficiency
When the tumors reached approximately 50–100 mm3, the mice were randomly divided into eight groups of six mice (day 0). The mice in group 1 to 8 were intravenously injected for each time with saline (200 μL), free Dox (4 mg kg−1), PFBT–Lip–FA (1 mg kg−1), Dio–Lip–FA (1 mg kg−1), Dox–Lip–FA (4 mg kg−1) with or without mild hyperthermia, and Dox–PFBT–Lip–FA (1 mg kg−1 of PFBT, 4 mg kg−1 of Dox) with or without mild hyperthermia, respectively. Each mouse received the tail vein injection every 2 days (a total of four times). The mice treated with mild hyperthermia were carried out the same as before. The tumor volume was measured every 2 days and calculated based on the equation: V = (a × b2)/2, where a and b were the long and short diameters of the tumor, respectively. The animals were also weighed at regular intervals during the experimental period.Histopathology analysis
At the 16th day after the first injection, the mice were sacrificed. Tumors and major organs were collected and fixed in 10% neutral buffered formalin. Sections of heart, liver, spleen, kidney, and lung were paraffin-embedded, nominally cut at 5 μm thickness, stained with hematoxylin and eosin, and examined by a board certified veterinary pathologist.Statistics analysis
All the experiments were repeated at least three times. Data were presented as means standard deviation (SD). One-way analysis of variance (ANOVA) was performed to determine significance among groups. All statistics were analyzed by two-tailed Student's t-test. p-Values less than 0.05 and 0.01 were considered to be statistically significant and highly significant, respectively.Results and discussion
Characterization of test liposomes
Four types of liposomes including Dox–Lip–FA, PFBT–Lip–FA, Dio–Lip–FA and PFBT-Dox–Lip–FA were fabricated via the thin film hydration method. The average diameter, PDI and zeta potential were summarized in Table 1. From the DLS determination, the size of the drug loaded or fluoprobes embedded liposomes was around 100 nm with the value of PDI < 0.22, demonstrating the uniform distribution of the liposomes. There were no significant differences (p > 0.05) among these test formulations in the vesicle size. In addition, all liposome systems displayed negative zeta potential, which was common for DSPE-PEG containing liposomes due to the shielded negatively charged phosphate group of DSPE-PEG.30 What's more, nanometer-scale associations could be formed when the alkyl tail length of the phospholipid was comparable to the alkyl side chain length of the conjugated polymer.31 When PFBT was added into the formulation, nanometer-scale association was formed between the phospholipid and the conjugated polymer. The bilayer of PFBT–Dox–Lip–FA was more compact than Dox–Lip–FA. Therefore, EE% values for PFBT–Dox–Lip–FA were significantly higher than those of Dox–Lip–FA (Table 1).| Formulation | Size (nm) | PDI | Zeta potential (mV) | EE% (Dox) |
|---|---|---|---|---|
| a Note: mean of three independent experiments. PDI: polydispersity index. EE%: encapsulation efficiency (%). Values are the means ± SD (n = 3). **p < 0.01. | ||||
| Dox–Lip–FA | 98.56 ± 2.40 | 0.21 ± 0.01 | −6.82 ± 0.70 | 70.2 ± 5.3 |
| PFBT–Lip–FA | 108.70 ± 2.10 | 0.19 ± 0.01 | −20.20 ± 1.10 | — |
| Dio–Lip–FA | 102.30 ± 1.80 | 0.19 ± 0.03 | −15.30 ± 1.40 | — |
| PFBT–Dox–Lip–FA | 127.30 ± 3.20 | 0.17 ± 0.01 | −25.00 ± 2.00 | 89.5 ± 4.6** |
TEM images of all tested formulations were shown in Fig. 1A–D, which was consistent with the DLS result. AFM was further used to confirm the particle surface morphology of the liposomes modified with FA ligand (as shown in Fig. S1A and B†). The results suggested the surface of non-targeted liposomes (PFBT–Dox–Lip) was relatively smooth, while the FA-targeted liposomes had small globular protrusions, which really verified the successful modification of liposomes with FA ligand.44
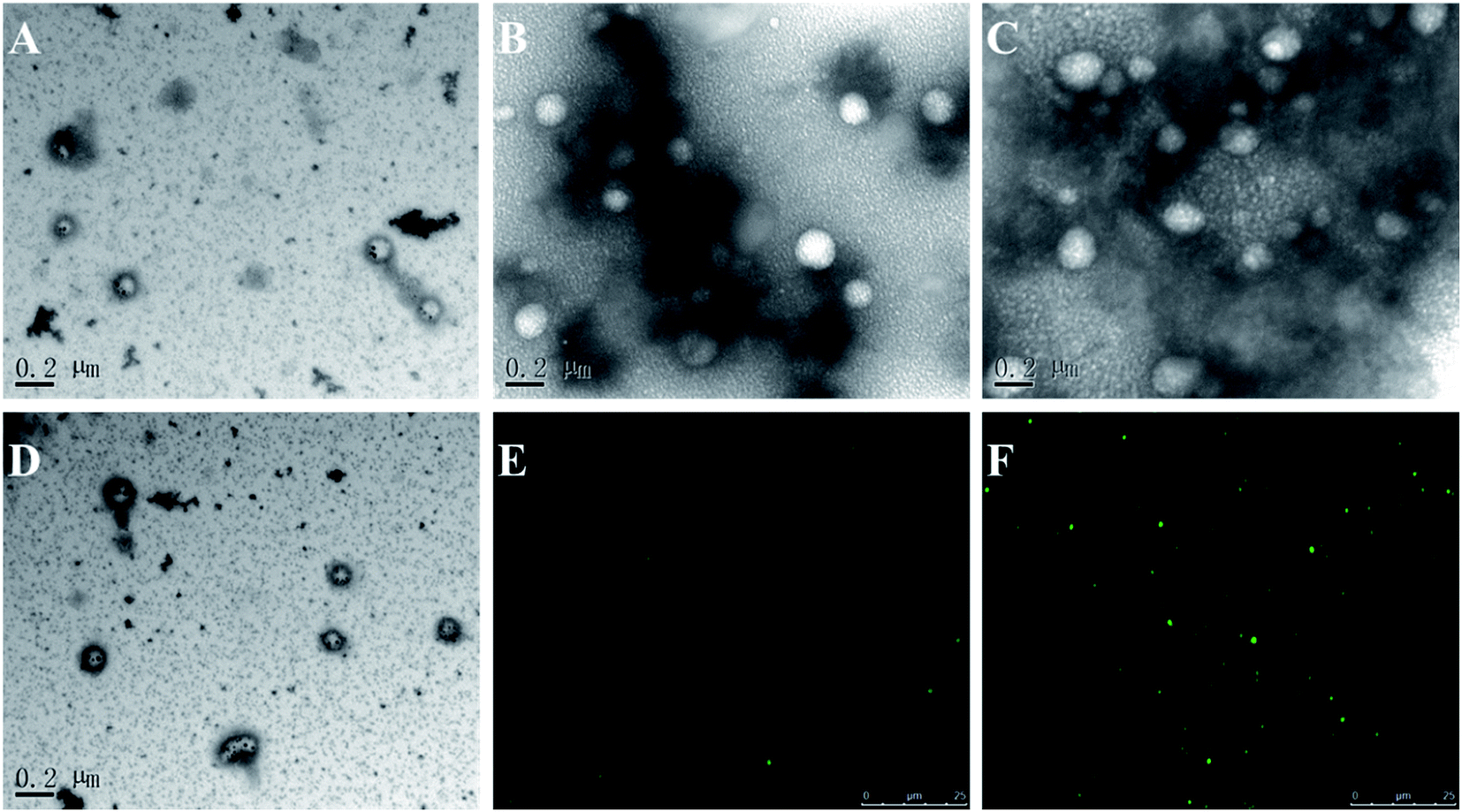 | ||
| Fig. 1 Transmission electron microscopy analysis of Dox–Lip–FA (A), Dio–Lip–FA (B), PFBT–Lip–FA (C) and PFBT–Dox–Lip–FA (D). Morphology of Dio–Lip–FA (E) and PFBT–Lip–FA (F) observed by confocal laser scanning microscope. Image A–D share the same scale bar of 0.2 μm. Image E and F share the same scale bar of 25 um. | ||
In addition, UV-vis absorption was carried out to confirm the incorporation of PFBT. As shown in Fig. 2A, the absorption peak of free PFBT was around 460 nm, while the peak was disappeared once it was entrapped into the bilayers. Superb fluorescence stability of the probes in biological environment was critical for their effectiveness applications in vivo. The thermal-stability of PFBT–Lip–FA was performed upon incubation the liposomes in phosphate-buffered saline buffer at 37 °C, using commercially available organic dyes Dio as reference. As shown in Fig. 2B, no obvious decrease in fluorescence intensity of PFBT–Lip–FA was observed in 7 days. In comparison, Dio–Lip–FA showed ∼23% decrease in fluorescence intensity under the same condition which revealed that the PFBT–Lip–FA possessed better thermal stability as compared to the widely utilized organic dyes. This was just consistent with a previous study in which the stability of conjugated polymer in vesicles was significantly better than organic dyes.32 Most importantly, as compared to Dio–Lip–FA, PFBT–Lip–FA showed higher brightness in the CLSM study (Fig. 1E and F) which indicated that PFBT was an ideal fluoprobe for theranostic liposomes.
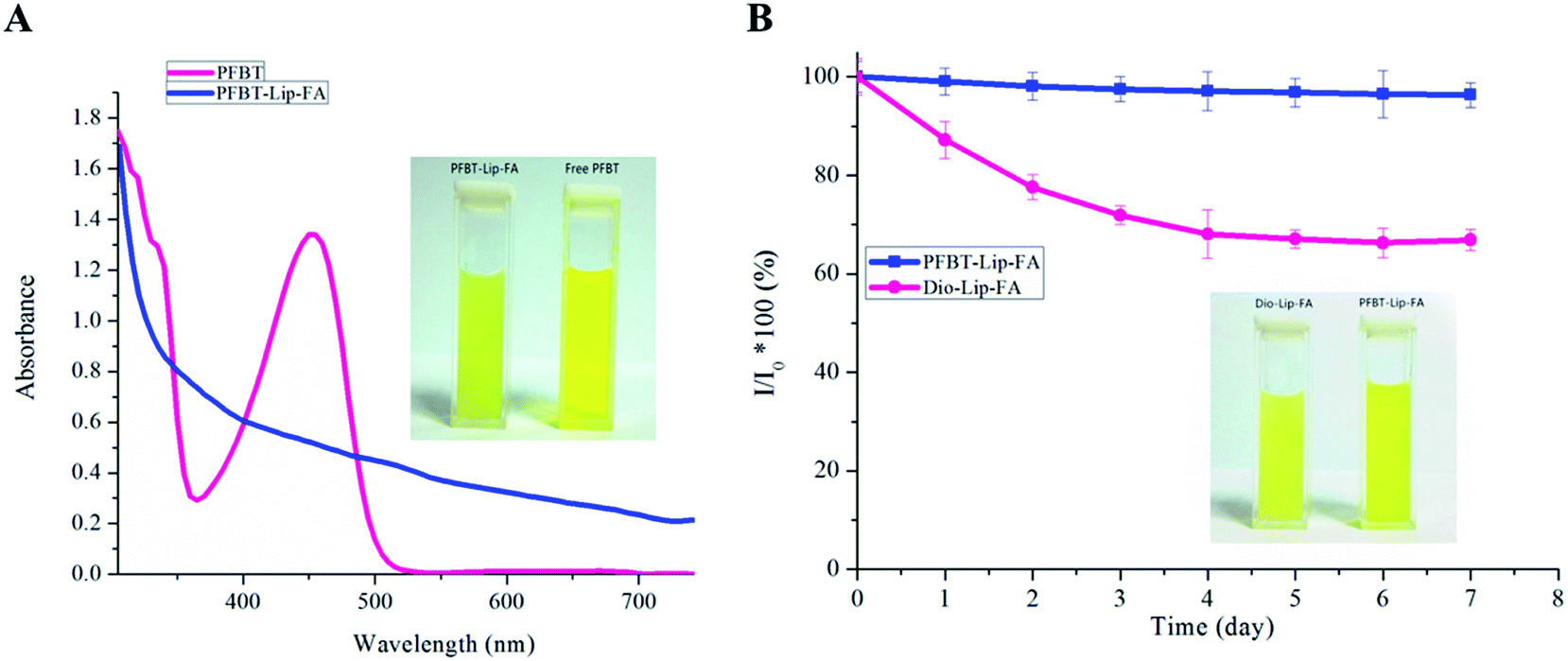 | ||
| Fig. 2 (A) UV-vis absorption spectra of the PFBT–Lip–FA and free PFBT. (B) Fluorescence intensity changes of Dio–Lip–FA and PFBT–Lip–FA upon incubation in PBS buffer at 37 °C for 7 days. | ||
In vitro drug release
To test thermo-sensitivity and release-kinetics of the developed Dox–Lip–FA, temperature- and time-dependent release profiles were determined, respectively. The release profile of Dox from Dox–Lip–FA at different temperatures (37–42 °C) was depicted in Fig. 3A. Rapid drug release was obtained by Dox–Lip–FA from 40 °C, but increased slightly when the temperature was over 41 °C. DPPC, with the transition temperature of 41.5 °C,33 was chosen as the primary constituent of these thermo-sensitive liposomes. As we all know, drug release occurred predominantly by the melting of lipids constituting the membrane.34 Thus temperature point of 42 °C was selected for the following release studies.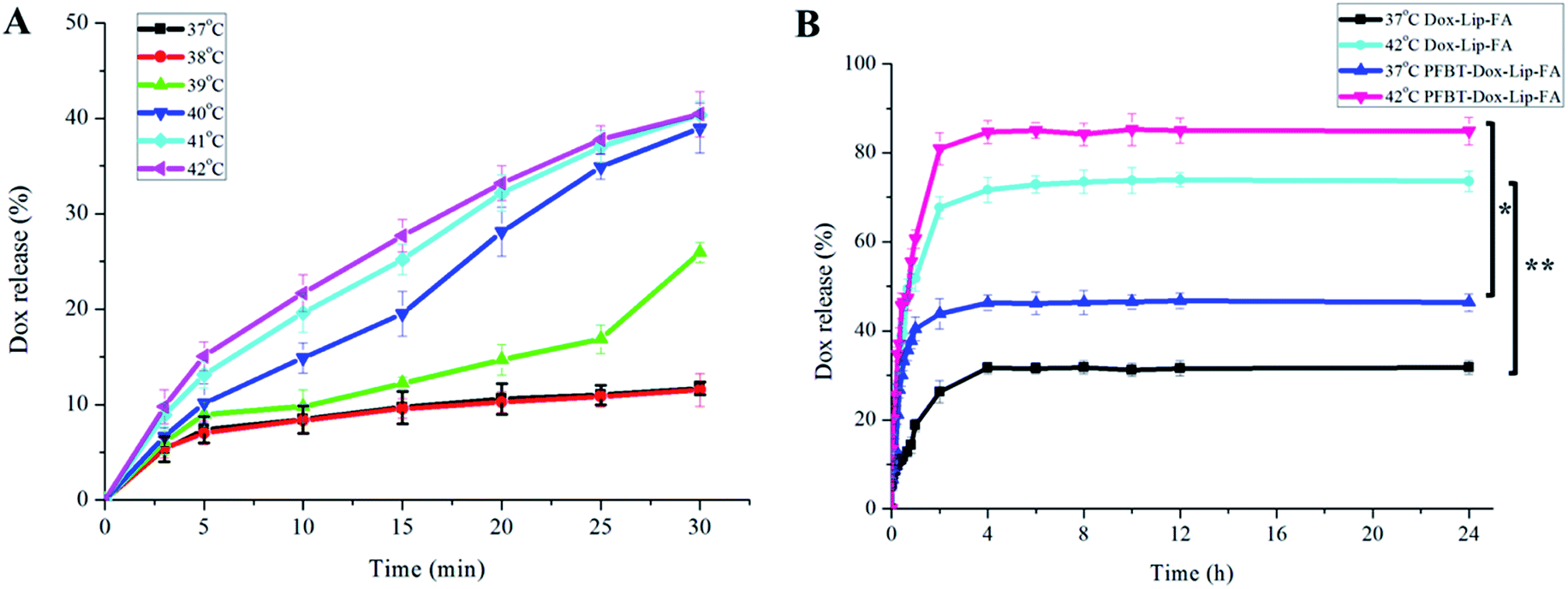 | ||
| Fig. 3 (A) Release profiles of drug from Dox–Lip–FA in PBS (pH 7.4) at different temperatures. (B) Release profiles of drug from both Dox–Lip–FA and PFBT–Dox–Lip–FA in 24 h at different temperatures. Data represent the mean ± standard deviation of at three experimental determinations (*p < 0.05; **p < 0.01). | ||
Then at the optimal drug release temperature of 42 °C, we followed release rate in time. Dox release at 37 °C was used as control. Fig. 3B showed the drug release profiles of both PFBT–Dox–Lip–FA and Dox–Lip–FA. Drug release from both formulations at 37 °C was very slow, with only 46.35% and 31.78% after 24 h, respectively. While at 42 °C, the Dox release rate was much faster, with the significant higher release approximately 84.87% and 73.62% within 24 h, respectively. In previous study, we have detected that the EE% of Dox in PFBT–Dox–Lip–FA was significantly higher than Dox–Lip–FA. When PFBT–Dox–Lip–FA was dialyzed, large amount of Dox was released from liposomes, and transported into the medium by the help of the high trans-membrane drug concentration gradient.35 Hence the drug release from PFBT–Dox–Lip–FA was enhanced as compared with Dox–Lip–FA at the same condition.
In vitro cytotoxicity
The cytotoxicity of the PFBT–Lip–FA to HepG2 and MCF-7 cells in comparison with Dio–Lip–FA was determined by cell survival to evaluate the biosafety of the liposomes. As shown in Fig. 4A and C, compared with the significantly reduced cell survival of Dio–Lip–FA treated cells, all the PFBT–Lip–FA groups did not introduce considerable cytotoxicity to the cells at the working concentration. Thus, the cytotoxicity of theranostic liposomes could mainly be attributed to the free Dox released from the acidic endosome after FA receptor-mediated endocytosis of the liposomes.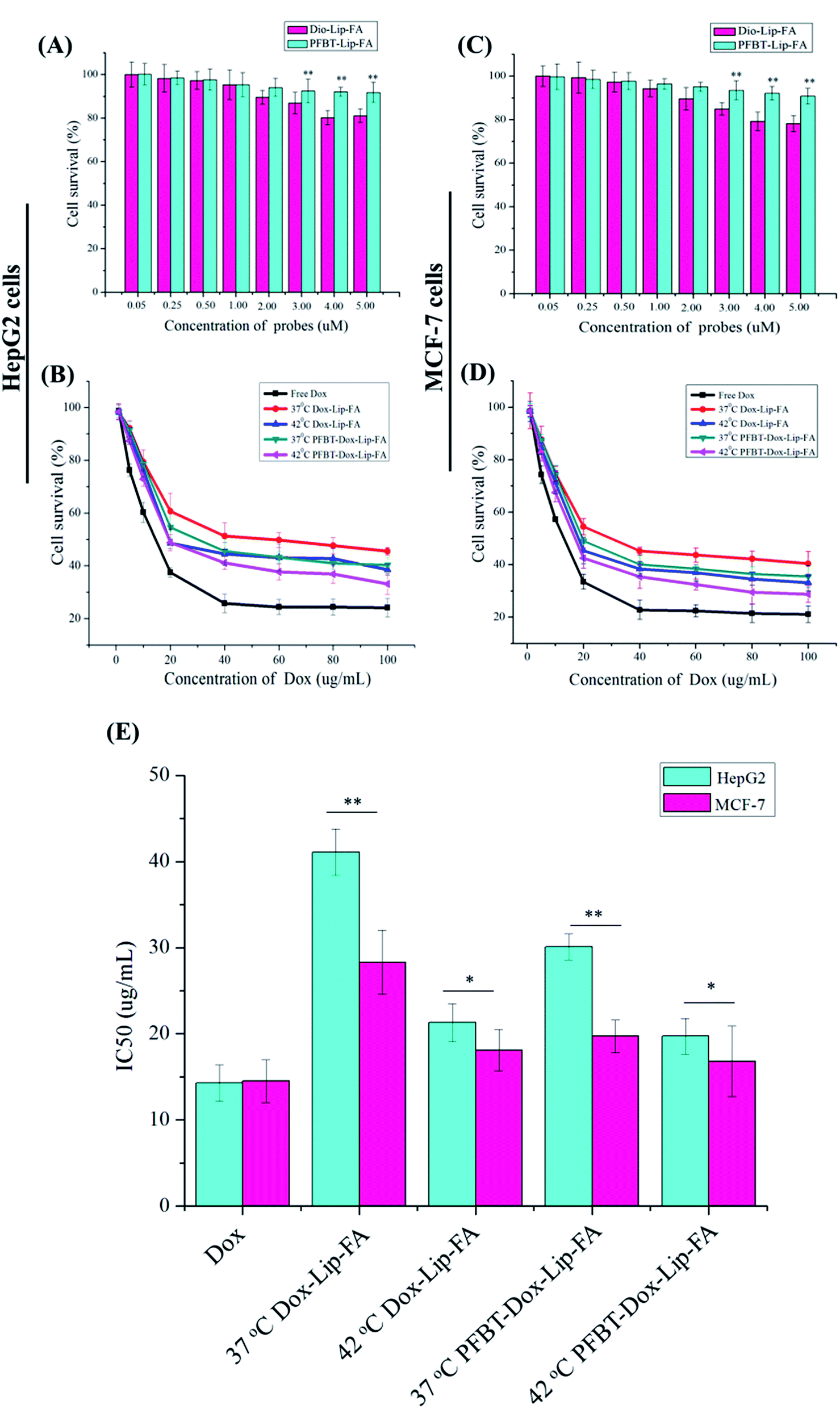 | ||
| Fig. 4 The cytotoxicity of PFBT–Lip–FA and Dio–Lip–FA against HepG2 (A) and MCF-7 (C) cells following 24 h incubation. The dose-dependent cell viability of HepG2 (B) and MCF-7 (D) cells following 24 h incubation with Dox-loaded samples and free Dox. The sample concentration ranged from 0 to 100 μg mL−1. The IC50 of PFBT–Dox–Lip–FA and Dox–Lip–FA liposomes in two types of cancer cell lines (E). Free Dox was used as the control and the liposome concentration was Dox-equivalent. Data are expressed as mean ± SD, n = 3 (*p < 0.05, **p < 0.01). | ||
Then the cytotoxicity of both PFBT–Dox–Lip–FA and Dox–Lip–FA at different temperature was evaluated with free Dox as the positive control. Fig. 4B and D demonstrated the survival rates of free Dox, PFBT–Dox–Lip–FA and Dox–Lip–FA under a series of Dox concentrations against HepG2 and MCF-7 cells, respectively. These results indicated that free Dox, PFBT–Dox–Lip–FA and Dox–Lip–FA could inhibit both types of cells' growth in a dose-dependent manner. Free Dox exhibited stronger cytotoxicity compared with PFBT–Dox–Lip–FA and Dox–Lip–FA. Under the in vitro condition, free Dox could be swiftly delivered into cells by passive diffusion and instantly inhibited cell growth.
On the other hand, cytotoxicity towards HepG2 and MCF-7 cells was compared in Fig. 4E. Significant greater cytotoxicity to MCF-7 cells was shown by PFBT–Dox–Lip–FA than to HepG2 cells. This might have been due to the specific binding between FA ligand and the FA receptor in MCF-7 cells through a receptor-mediated active targeting, demonstrating the advantage of PFBT–Dox–Lip–FA for tumor treatment. More importantly, the IC50 of PFBT–Dox–Lip–FA to MCF-7 cells was 19.7 ± 1.9 μg mL−1 at 37 °C and 16.8 ± 4.5 μg mL−1 at 42 °C, whilst the corresponding IC50 of Dox–Lip–FA was 28.3 ± 3.7 μg mL−1 at 37 °C and 18.1 ± 2.4 μg mL−1 at 42 °C, respectively. As previously reported, maximum therapeutic effects might only be achieved when tumor cells were subjected to a maximum drug exposure.36 Therefore, compared with Dox–Lip–FA, PFBT–Dox–Lip–FA exhibited obviously inherent cytotoxicity to the cancer cells, which could be attributed to the larger amount of drug released in the culture medium (consistent with the in vitro drug release) and then diffused into tumor cells during the incubation period.
Cellular uptake
In order to clarify the internalization of the liposomes, the cell nuclei were stained with DAPI (blue) while the liposomes were labeled with PFBT/Dio (green) and Dox (red). As shown in Fig. 5, the significantly strong green fluorescence in cytoplasm of PFBT–Dox–Lip–FA treated both HepG2 and MCF-7 cells revealed the higher brightness of PFBT to Dio, which was consistent with the previous characterization study.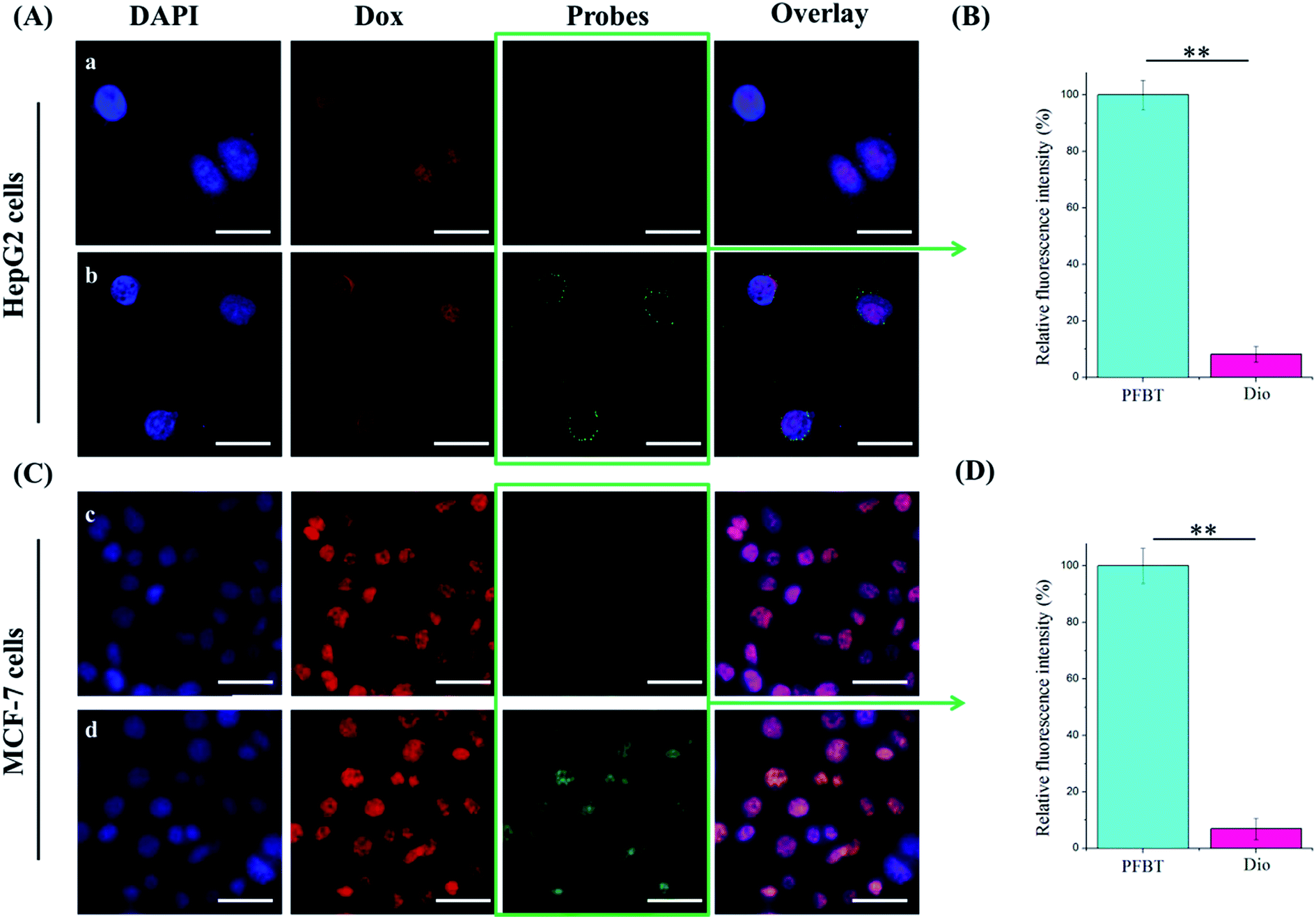 | ||
| Fig. 5 Confocal laser scanning microscopic images of HepG2 (A) and MCF-7 (C) cells after incubating with different fluoprobes labeled Dox–Lip–FA for 1 h. Dox in red and fluoprobes (PFBT or Dio) in green. Relative fluorescence intensity of PFBT or Dio emissions from HepG2 (B) and MCF-7 (D) cells in images (*p < 0.05, **p < 0.01). Scale bar: 25 μm (A), 50 μm (C). | ||
Fig. 6A and B indicated the accumulation of drugs and fluoprobes in the two cell lines, respectively. Nuclear co-localization of Dox and DAPI was observed by treated with PFBT–Dox–Lip–FA. Moreover, the fluorescence intensity of Dox was obviously improved with the increased temperature or time, which was most likely due to the higher drug accumulation in tumor cells. Considering only the free drug that reached the nuclei of the tumor cells could ensure the DNA intercalation,37,46 which was attributed to the antiproliferative activity of Dox, PFBT–Dox–Lip–FA showed better antiproliferative activity in the cytotoxicity study.
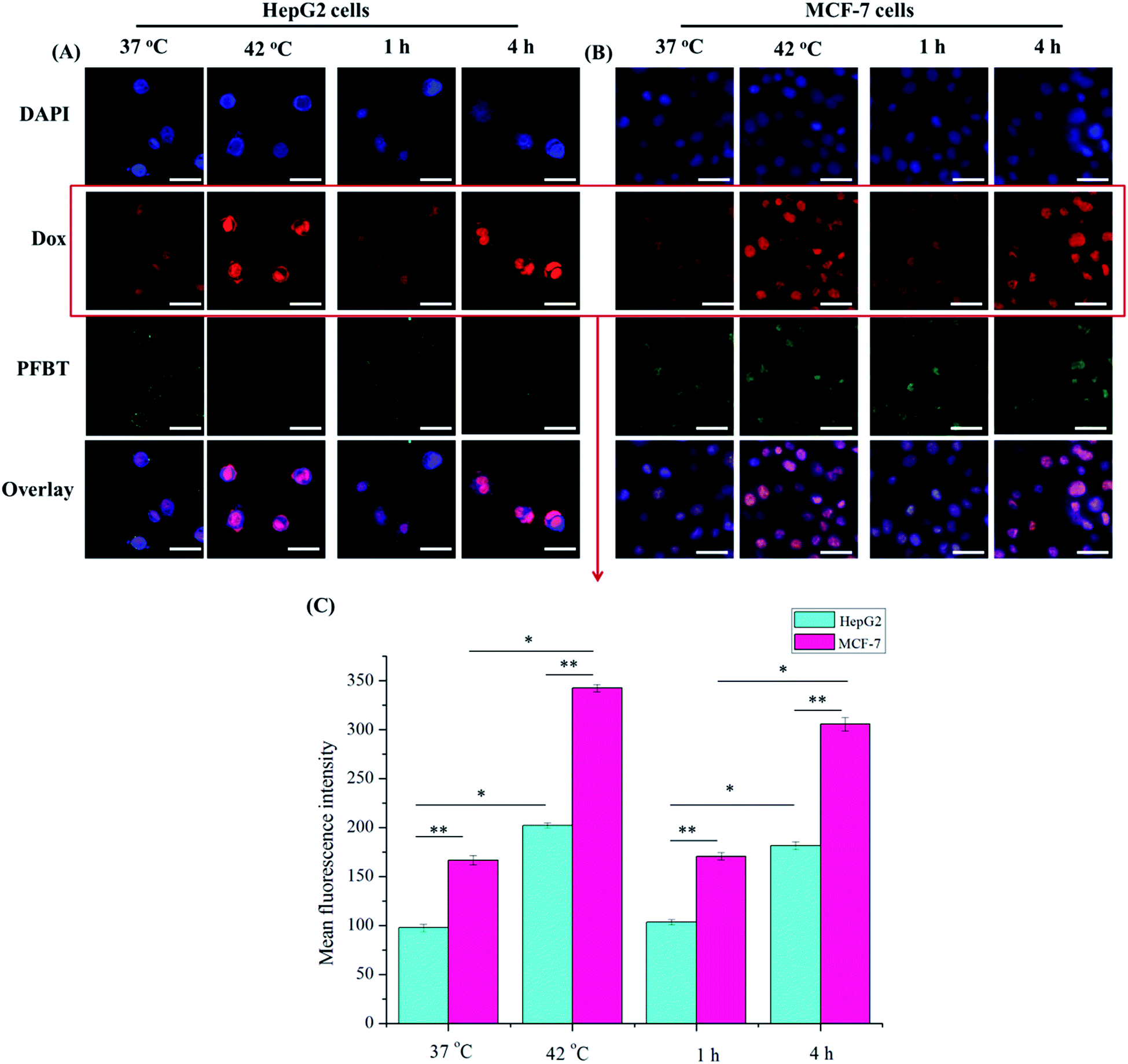 | ||
| Fig. 6 Confocal laser scanning microscopic images of HepG2 (A) and MCF-7 (B) cells after incubating with PFBT–Dox–Lip–FA for different temperatures (37 °C and 42 °C, 1 h) or times (1 h and 4 h, 37 °C). Dox in red and fluoprobes (PFBT or Dio) in green. (C) Mean fluorescence intensity of Dox emissions from cells in images. (*p < 0.05, **p < 0.01). Scale bar: 25 μm (A), 50 μm (B). | ||
FA was able to be efficiently internalized into the cells through the receptor mediated endocytosis even when conjugated with a wide variety of molecules.45 As shown in Fig. 6, significantly stronger red fluorescence was detected in nuclei of PFBT–Dox–Lip–FA treating MCF-7 cells at different temperature (compared to those of HepG2 cells), which indicated the improvement of intracellular uptake of Dox. Similar phenomenon could be also observed in the time-dependent cellular uptake study. These results indicated that MCF-7 cells effectively internalized the PFBT–Dox–Lip–FA by FR mediation. Therefore, within the same dosage, the optimal liposomes could more accurately deliver the drug to the MCF-7 cells, thus reducing side effects.
In vivo imaging
The promising results in cell imaging inspired us to further explore the feasibility of PFBT–Dox–Lip–FA as an in vivo theranostic delivery system. To estimate the in vivo liposomal delivery to tumors, MCF-7 tumor-bearing nude mice were established. The fluorescence intensity of tumors was detected after injection of different formulations for 6 h (Fig. 7A). Unfortunately, only autofluorescence of the mice could be detected in the groups of Dox–Lip–FA and Dio–Lip–FA due to the emission wavelength of Dox (580 nm) and Dio (501 nm). Indeed, in this portion of the electromagnetic spectrum the absorption of biomolecules (water, hemoglobin, etc.), the autofluorescence of the cells, and the light scattering were high, resulting in limiting tissue penetration.38 While PFBT had an identical maximum at 598 nm (excitation at 440 nm), which was highly desirable, since it minimized the disturbance of autofluorescence from biological substances.39 More importantly, as compared to Dox–Lip–FA, PFBT–Lip–FA showed higher brightness in the fluorescence study (Fig. S2†) which also induced better detection of PFBT in live animals. As shown in Fig. 7A, PFBT–Lip–FA was identified to be mainly accumulated in the tumor site. Therefore, PFBT could be used as a fluorescent marker for imaging-guided chemotherapy.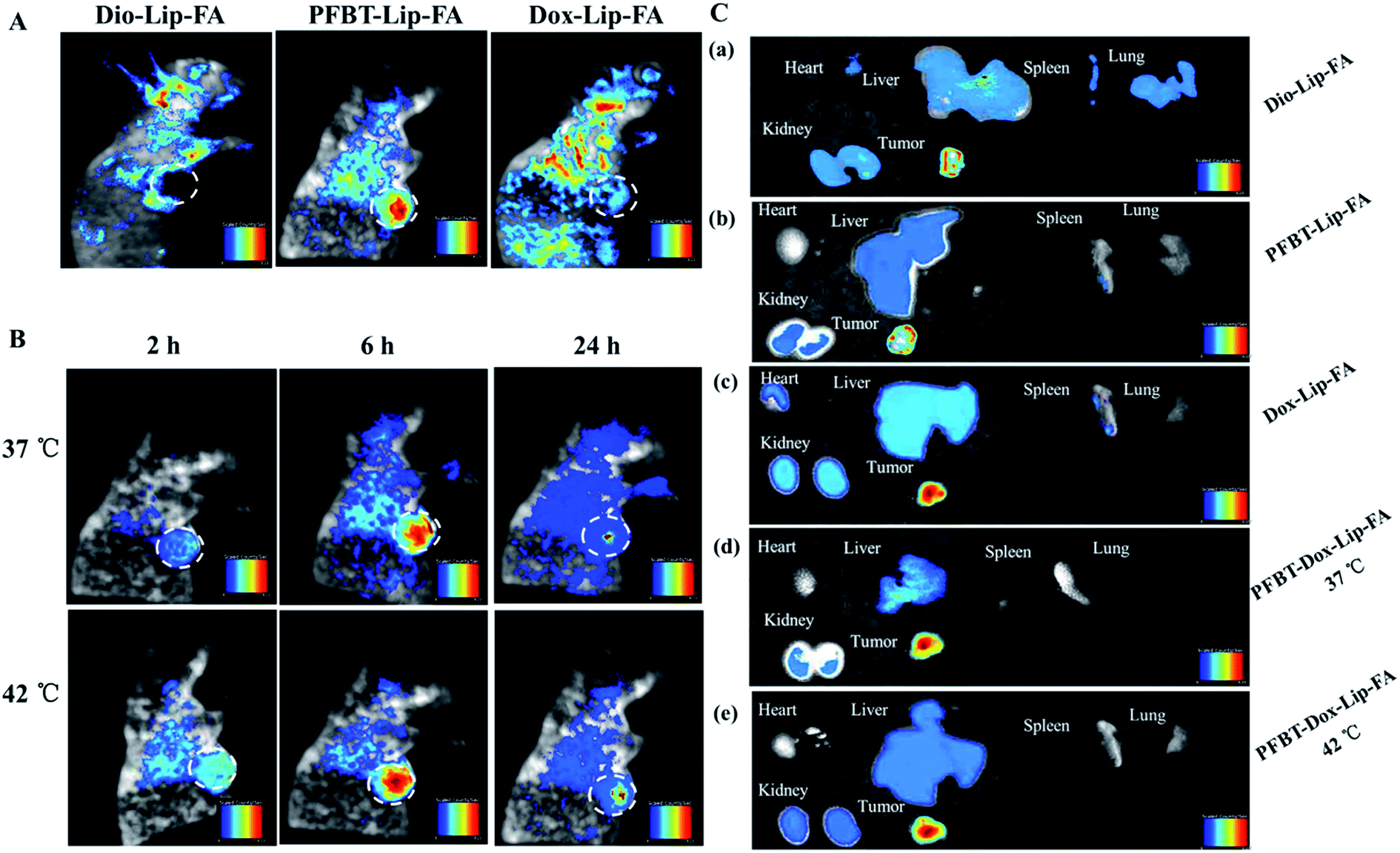 | ||
| Fig. 7 In vivo imaging and biodistribution of nude mice bearing MCF-7 tumors after venous injection of different prepared liposomes. (A) Prescription-dependent fluorescent images of nude mice. (B) Time-lapse fluorescent images of nude mice after injecting PFBT–Dox–Lip–FA in vein. (C) Ex vivo fluorescent image of tumors and organs after 24 h circulation in the body. | ||
Fig. 7B showed the fluorescence signal and intensity distribution as a function of time for theranostic liposomes delivered systemically via tail vein injections at 37 °C and 42 °C, respectively. It was clear that there was an increased accumulation of PFBT–Dox–Lip–FA in tumor site during the first 6 h, and subsequently decreased gradually. Hyperthermia was a well-established method to augment liposomal accumulation into solid tumors.40 It also has been demonstrated that enhancement of tumor accumulation by hyperthermia was due to increases in local blood flow,41 a reported 40–60% increase at 42 °C, resulting in increased microvascular permeability.42 Thus accumulation of theranostic liposomes with mild hyperthermia in the tumor tissue was significantly higher than that without mild hyperthermia. At 24 h time point post-injection, the liposomes retained detectable signal in the tumor then. PFBT–Dox–Lip–FA clearly displayed the tumor against surrounding tissues, showing its high efficiency of tumor imaging.
The fluorescent intensity of various tissues after 24 h injection was also studied (Fig. 7C). Qualitatively, Dio fluorescence intensity in all organs including heart, liver, spleen, lungs, and kidneys, with the exception of tumor, was much higher than PFBT–Lip–FA and PFBT–Dox–Lip–FA group (Fig. 7C). In the case of administration with Dox–Lip–FA, the fluorescence images of the organs showed specific Dox accumulation in tumor tissue as well as in the heart, liver and kidney. Most importantly, the lower accumulation of PFBT–Dox–Lip–FA in hearts should also be noticed, which might reduce the cardiotoxicity of Dox due to the liposome delivery systems. These results collectively demonstrated that PFBT–Dox–Lip–FA could help Dox specifically accumulated in tumors, thus suit for delivering anticancer drugs to cancer cells in vivo. In addition, it also proved that PFBT–Dox–Lip–FA was a desired carrier for simultaneously diagnose and targeting drug delivery.
In vivo antitumor efficacy
On the basis of the above results, the in vivo antitumor efficacy and systemic toxicity of the dual loaded liposomes were further investigated on human liver tumor-bearing nude mice. As shown in Fig. 8A, the tumors treated with PBS, Dio–Lip–FA and PFBT–Lip–FA showed rapid growth, suggesting that the fluoprobes embedded liposomes did not inhibit the tumor growth. While the tumors in all drug-treated groups have shown more obvious growth retardation since the second treatment compared to control. In particularly, PFBT–Dox–Lip–FA combined with mild hyperthermia exhibited the highest inhibitory effects on tumor growth during the whole test time (up to 16 d), with the smallest tumor sizes (298.1 ± 38.1 mm3) at the end of the study. The superior antitumor effect of theranostic liposomes might be attributed to the enhanced liposomal accumulation during the blood circulation, the higher drug amount delivery to the tumor site, and the efficient cellular uptake in the tumor tissue. By the end of the study, the excised tumors after the treatment were dissected and photographed (Fig. 8A).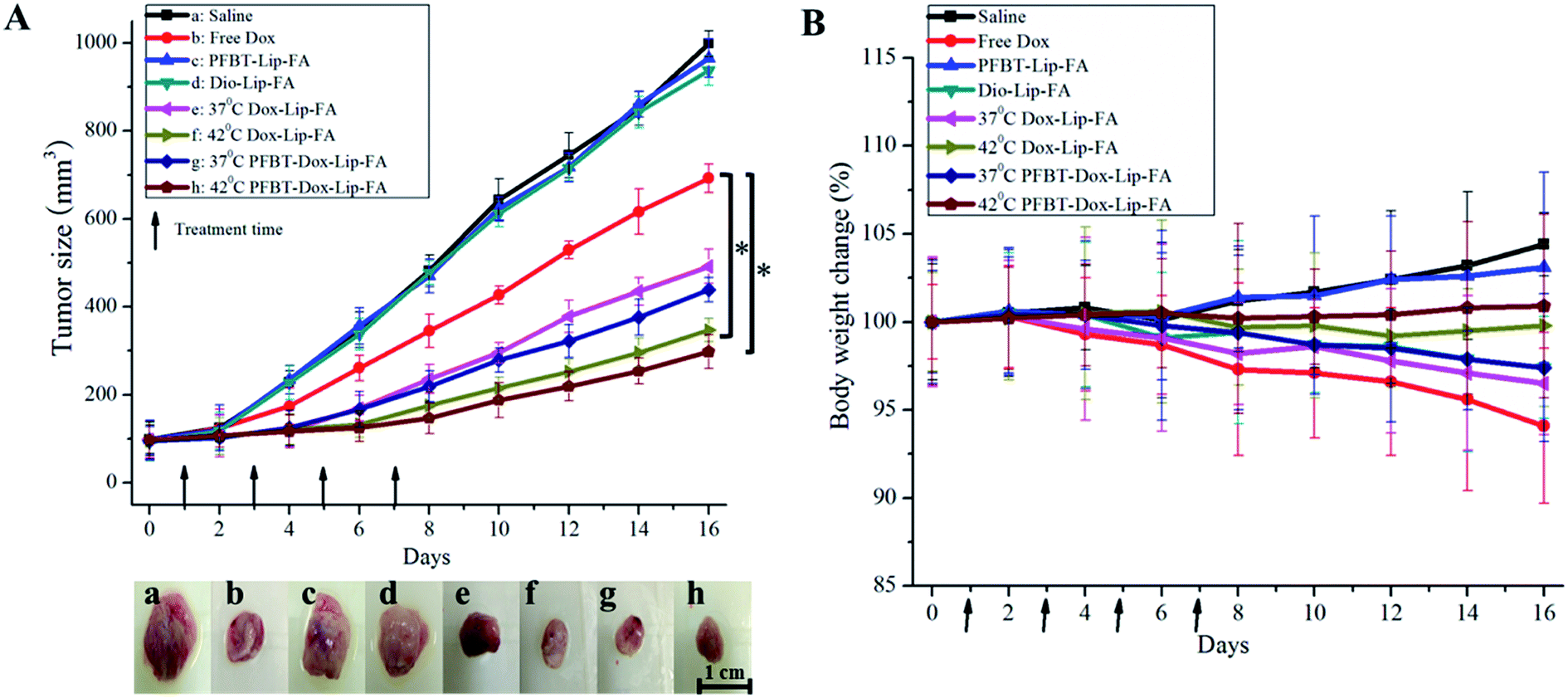 | ||
| Fig. 8 Tumor volume and body weight changes after anticancer treatment with different formulations in mice bearing MCF-7 human breast cancer xenograft. (A) Growth curves of MCF-7 tumors in nude mice that were treated with different formulations and digital photographs of tumors on day 16. Arrows indicated days on which mice were treated (*p < 0.05). (B) Body weight changes were recorded during the treatment. Arrows indicated days on which mice were treated. | ||
Simultaneously, the body weights of mice during the treatment period were also monitored to investigate the adverse effects of various formulations.43 As shown in Fig. 8B, the body weights of PBS and PFBT–Lip–FA treated mice showed continuous and slow increase, which might be partly ascribed to the tumor growth and low toxicity of PFBT at the dose of 1 mg kg−1. Obvious weight loss (about 6%) was observed in mice treated with free Dox at 4 mg kg−1, indicating the potential of side effects. On the contrary, the treatment with theranostic liposomes combined with mild hyperthermia did not lead to any significant body weight loss, which might be due to their higher accumulation in tumor site and lower distribution in normal organs. With the high antitumor efficacy and the low drug-related toxicity, the theranostic liposomes were promising in cancer diagnose and targeting therapy.
Histopathology analysis
To further investigate the antitumor efficacy and in vivo toxicity of test liposomes, histological studies were employed to detect cancerous metastases and induced toxicity. After 16 days of treatments, the tumors and main organs of the post-treated mice were removed, and the cell proliferation and apoptosis in the tumors of each group were evaluated by hematoxylin and eosin studies. As shown in Fig. 9F, similar to the PBS group (Fig. 9A), no noticeable signal of organ damage was observed from the stained organ slices of the PFBT–Lip–FA treated mice. In contrast, significant pathological changes in liver and lung (Fig. 9E, black arrows) of mice were observed for Dio–Lip–FA group, which was consistent with the results of biodistribution study. It was notable that, hepatotoxicity was found (Fig. 9C and D, black arrows) for Dox–Lip–FA treated group. However, organs of PFBT–Dox–Lip–FA treated group were normal without any visible difference, suggesting the biosafety of the optimal formulation (Fig. 9G and H).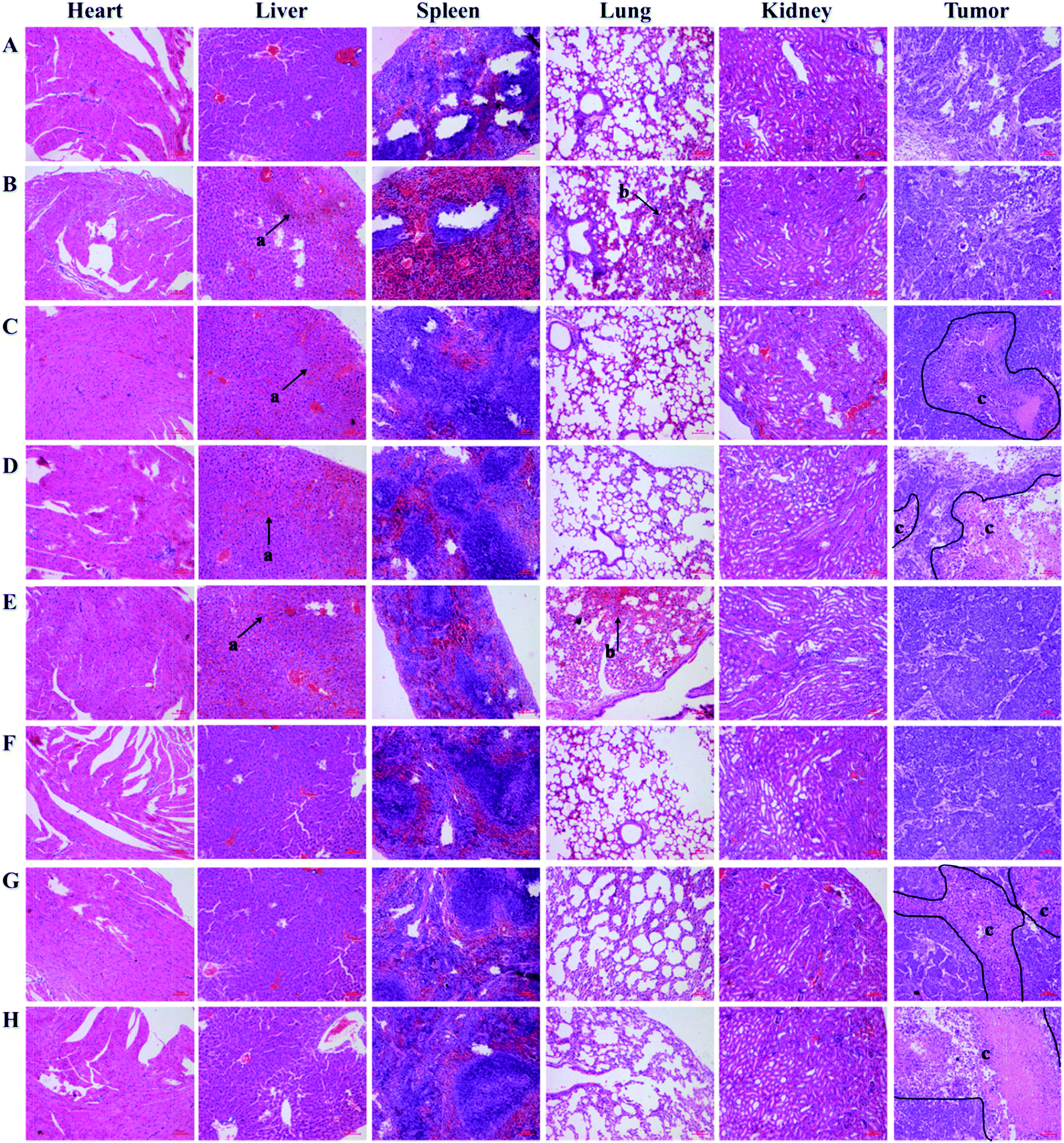 | ||
| Fig. 9 Microphotographs of heart, liver, spleen, lung, kidney and tumor from mice in various treatment groups stained with H&E ((a) inflammation necrotic areas, (b) hemorrhagic regions, and (c) necrotic and apoptosis areas). Saline (A), free Dox (B), Dox–Lip–FA 37 °C (C), Dox–Lip–FA 42 °C (D), Dio–Lip–FA (E), PFBT–Lip–FA (F), PFBT–Dox–Lip–FA 37 °C (G), PFBT–Dox–Lip–FA 42 °C (H). Bar, 20 μm. | ||
Tumors in mice that were injected with sterile PBS (i.e., negative control) showed large cell nuclei and small intercellular spaces, but rarely observable of necrotic and apoptosis. Besides, tumors of free Dox treated mice had some necrotic areas and small apoptosis regions. In contrast, numerous necrotic and apoptosis areas (Fig. 9G and H, inside the black line) were observed by tumors of Dox–Lip–FA and PFBT–Dox–Lip–FA treated group. Considering therapeutic efficacy and toxicity, PFBT–Dox–Lip–FA was a safe and effective vehicle for cancer imaging and targeting therapy.
Conclusions
In conclusion, we have developed a novel theranostic platform based on PFBT-embedded and Dox-loaded liposomes for cancer imaging and targeting therapy in nude mice. As compared to commercially available organic dyes Dio, PFBT–Lip–FA has shown better thermo-stabilities, higher brightness and lower toxicity. In addition, PFBT–Dox–Lip–FA was superior to Dox–Lip–FA in terms of drug release, cytotoxicity, cellular uptake and in vivo tumor targeting accumulation. Overall, the introduction of PFBT in the liposomes not only facilitated the cancer imaging in nude mice, but also improved the drug accumulation in the tumor site. The current work highlighted the significance of theranostic liposomes in delivering therapeutic drugs and diagnostic imaging agents at the same time. By encapsulating various drugs into the theranostic liposomes will easily broaden the use of conjugated polymer-based multifunctional liposomes for potential clinical applications.Conflict of interest
The authors declare no conflict of interests.Acknowledgements
We thank Dan Ding for kindly providing conjugated polymer dots of PFBT. This work is supported by Tianjin Research Program of Application Foundation and Advanced Technology (15JCQNJC13800), National Natural Science Foundation of China (81503016) and National “973” Program of China (2014CB932200).References
- R. Siegel, D. Naishadham and A. Jemal, Ca-Cancer J. Clin., 2012, 62(1), 10–29 CrossRef PubMed.
- P. P. Shanbhag, S. V. Jog and M. M. Chogale, et al., Curr. Drug Delivery, 2013, 10(3), 357–362 CrossRef.
- B. T. Luk, R. H. Fang and L. Zhang, Theranostics, 2012, 2, 1117–1126 CrossRef CAS PubMed.
- K. Y. Choi, G. Liu and S. Lee, et al., Nanoscale, 2012, 4(2), 330–342 RSC.
- Y. Urano, D. Asanuma and Y. Hama, et al., Nat. Med., 2009, 15(1), 104–109 CrossRef CAS PubMed.
- C. Richard, Q. M. Chermont and D. Scherman, Tumori, 2008, 94(2), 264 CAS.
- W. Wu, R. Li and X. Bian, et al., ACS Nano, 2009, 3(9), 2740–2750 CrossRef CAS PubMed.
- T. Nam, S. Park and S. Y. Lee, et al., Bioconjugate Chem., 2010, 21(4), 578–582 CrossRef CAS PubMed.
- Y. Waerzeggers, P. Monfared and T. Viel, et al., BBA, Mol. Basis Dis., 2010, 1802(10), 819–839 CrossRef CAS PubMed.
- J. H. Kim, Y. S. Kim and K. Park, et al., Biomaterials, 2008, 29(12), 1920–1930 CrossRef CAS PubMed.
- J. Zhang, R. E. Campbell and A. Y. Ting, et al., Nat. Rev. Mol. Cell Biol., 2002, 3(12), 906–918 CrossRef CAS PubMed.
- X. Gao, Y. Cui and R. M. Levenson, et al., Nat. Biotechnol., 2004, 22(8), 969–976 CrossRef CAS PubMed.
- A. de La Zerda, C. Zavaleta and S. Keren, et al., Nat. Nanotechnol., 2008, 3(9), 557–562 CrossRef CAS PubMed.
- W. J. M. Mulder, G. J. Strijkers and G. A. F. van Tilborg, et al., Acc. Chem. Res., 2009, 42(7), 904–914 CrossRef CAS PubMed.
- A. M. Smith, H. Duan and A. M. Mohs, et al., Adv. Drug Delivery Rev., 2008, 60(11), 1226–1240 CrossRef CAS PubMed.
- K. Li and B. Liu, J. Mater. Chem., 2012, 22(4), 1257–1264 RSC.
- D. Ding, K. Li and Z. Zhu, et al., Nanoscale, 2011, 3(5), 1997–2002 RSC.
- Y. Yuan, D. Ding and K. Li, et al., Small, 2014, 10(10), 1967–1975 CrossRef CAS PubMed.
- A. Duarte, K. Y. Pu and B. Liu, et al., Chem. Mater., 2010, 23(3), 501–515 CrossRef.
- C. Wu, B. Bull and C. Szymanski, et al., ACS Nano, 2008, 2(11), 2415–2423 CrossRef CAS PubMed.
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36(7), 887–913 CrossRef CAS.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52(11), 3086–3109 CrossRef CAS PubMed.
- A. Akbarzadeh, R. Rezaei-Sadabady and S. Davaran, et al., Nanoscale Res. Lett., 2013, 8(1), 102 CrossRef PubMed.
- R. T. Chacko, J. Ventura and J. Zhuang, et al., Adv. Drug Delivery Rev., 2012, 64(9), 836–851 CrossRef CAS PubMed.
- M. J. W. Johnston, S. C. Semple and S. K. Klimuk, et al., Biochim. Biophys. Acta, Biomembr., 2007, 1768(5), 1121–1127 CrossRef CAS PubMed.
- R. D. Hofheinz, S. U. Gnad-Vogt and U. Beyer, et al., Anti-Cancer Drugs, 2005, 16(7), 691–707 CrossRef CAS PubMed.
- Z. Wang, Y. Yu and W. Dai, et al., Biomaterials, 2012, 33(33), 8451–8460 CrossRef CAS PubMed.
- P. S. Low, W. A. Henne and D. D. Doorneweerd, Acc. Chem. Res., 2007, 41(1), 120–129 CrossRef PubMed.
- K. Kono, T. Ozawa and T. Yoshida, et al., Biomaterials, 2010, 31(27), 7096–7105 CrossRef CAS PubMed.
- Z. S. Al-Ahmady and K. Kostarelos, Biomaterials, 2012, 33(18), 4608–4617 CrossRef PubMed.
- J. Yoon, J. Kwag and T. J. Shin, et al., Adv. Mater., 2014, 26(26), 4559–4564 CrossRef CAS PubMed.
- D. Ding, J. Liu and G. Feng, et al., Small, 2013, 9(18), 3093–3102 CrossRef CAS PubMed.
- D. Needham, J. Y. Park and A. M. Wright, et al., Faraday Discuss., 2013, 161, 515–534 RSC.
- G. N. C. Chiu, S. A. Abraham and L. M. Ickenstein, et al., J. Controlled Release, 2005, 104(2), 271–288 CrossRef CAS PubMed.
- K. J. Chen, E. Y. Chaung and S. P. Wey, et al., ACS Nano, 2014, 7, 5105–5115 CrossRef PubMed.
- T. M. Allen, Nat. Rev. Cancer, 2002, 2, 750–763 CrossRef CAS PubMed.
- J. Shi, Y. Liu and L. Wang, et al., Acta Biomater., 2014, 10(3), 1280–1291 CrossRef CAS PubMed.
- C. G. Hadjipanayis, H. Jiang and D. W. Roberts, et al., Semin. Oncol., 2011, 38(1), 109–118 CrossRef PubMed.
- K. Y. Pu and B. Liu, Adv. Funct. Mater., 2009, 19(2), 277–284 CrossRef CAS.
- G. Kong and M. W. Dewhirst, Int. J. Hyperthermia, 1999, 15, 345–370 CrossRef CAS PubMed.
- T. Karino, S. Koga and M. Maeta, Jpn. J. Surg., 1988, 18, 276–283 CrossRef CAS PubMed.
- G. Kong, R. D. Braun and M. W. Dewhirst, Cancer Res., 2001, 61, 3027–3032 CAS.
- I. Klimtová, T. Šimůnek and Y. Mazurová, et al., Hum. Exp. Toxicol., 2002, 21(12), 649–657 Search PubMed.
- X. Ma, J. Zhou and C. X. Zhang, et al., Biomaterials, 2013, 34(18), 4452–4465 CrossRef CAS PubMed.
- J. M. Chan, L. Zhang and K. P. Yuet, et al., Biomaterials, 2009, 30(8), 1627–1634 CrossRef CAS PubMed.
- R. I. Pakunlu, Y. Wang and M. Saad, et al., J. Controlled Release, 2006, 114(2), 153–162 CrossRef CAS PubMed.
- S. Brahmachari, M. Ghosh and S. Dutta, et al., J. Mater. Chem. B, 2014, 2(9), 1160–1173 RSC.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65(1), 71–79 CrossRef CAS PubMed.
- S. Brahmachari, D. Das and A. Shome, et al., Angew. Chem., Int. Ed., 2011, 50(47), 11243–11247 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra24485d |
| This journal is © The Royal Society of Chemistry 2016 |