
Synthesis of 4-phenyl-5,6-dihydrobenzo[h]quinazolines and their evaluation as growth inhibitors of carcinoma cells†
Hardesh K. Maurya a,
Mohammad Hasanaind,
Sarita Singha,
Jayanta Sarkard,
Vijaya Dubeyb,
Aparna Shuklac,
Suaib Luqmanb,
Feroz Khanc and
Atul Gupta‡
*a
a,
Mohammad Hasanaind,
Sarita Singha,
Jayanta Sarkard,
Vijaya Dubeyb,
Aparna Shuklac,
Suaib Luqmanb,
Feroz Khanc and
Atul Gupta‡
*a
aMedicinal Chemistry Department, CSIR-Central Institute of Medicinal and Aromatic Plants, P.O. CIMAP, Kukrail Road, Lucknow 226015, India. E-mail: atisky2001@yahoo.co.in; Tel: +915222718556
bMolecular Bioprospection Department, CSIR-Central Institute of Medicinal and Aromatic Plants, P.O. CIMAP, Kukrail Road, Lucknow 226015, India
cMetabolic Structural Biology Department, CSIR-Central Institute of Medicinal and Aromatic Plants, P.O. CIMAP, Kukrail Road, Lucknow 226015, India
dDivision of Biochemistry, CSIR-Central Drug Research Institute, Sector-10, Jankipuram Extension, Lucknow 226 031, India
First published on 26th January 2016
Abstract
The synthesis of various benzo[h]quinazoline analogs (4a–f, 6a–d, 8a and 8b) was accomplished through the reaction of chalcone with guanidine. The synthesized compounds (4a–f, 6a–d, 8a and 8b) were screened for their anticancer potential against different cancer cells viz MCF-7, DLD1, A549, DU145 & FaDu cell lines. Compounds 4a, 6a–d & 8b showed significant anticancer activity in these cancer cell lines with a range of IC50 values of 1.5–12.99 μM. A functional study of promising molecule 6d at 7 μM (at the IC50 value) over 24 and 48 h showed that it possesses anticancer activity through triggering apoptosis. In a tubulin polymerization assay, 6d effectively inhibited tubulin polymerization with an IC50 of 2.27 μM. In silico docking studies of 6d revealed that 6d has good affinity with an estrogen receptor as well as a tubulin protein on its β-sheet of the colchicines binding site.
Introduction
Any defect in the programmed cell growth of a normal healthy cell can lead to an accumulation of undesired defective cells and ultimately result in the development of cancer. Cancer is a leading cause of death worldwide. The present statistics reveal that it accounts for one in every seven deaths globally which is more than for any infectious disease.1 By 2030, it is expected that about 21.7 million new cancer cases and 13.0 million cancer deaths will be reported.2 Lung, prostate, colorectum, stomach, and liver cancers are very common in males while in females stomach, breast, uterine and cervix cancers are frequent and the common cause of cancer is hormone imbalance.3 Among several other estrogen-dependent cancers, breast cancer is a leading pandemic disease that affects women over a wide age group.3 Considering the necessity of anticancer chemotherapeutics for the treatment of estrogen-dependent cancers such as breast cancer, different estrane based molecules such as fulvestrant (ICI-182,780, I), ICI-164,384 (II), SR-16157 (IV), SR-16234 (V), 3-hydroxy-estra-1,3,5(10)trien-17-oxo-15β-morpholinobutan-1-one (VI), estradiol-chlorambucil conjugates (VI), estra-13,5(10)-trien-16,17[3,4]-pyrazolo-N-(3-ehtylpyridino)-5-caboxamide (VII), VP-128 (VIII), and 16α,β-[11-(2-pyridylethylamino)undecanyl]-estradiol dichloroplatium (II) complexes (IX) have been discovered with potent anti-breast cancer potential, Fig. 1.4–11 It is reported that estrogen receptor-α (ER-α) dominates in breast cancer tissue and is the main culprit of excess cell proliferation.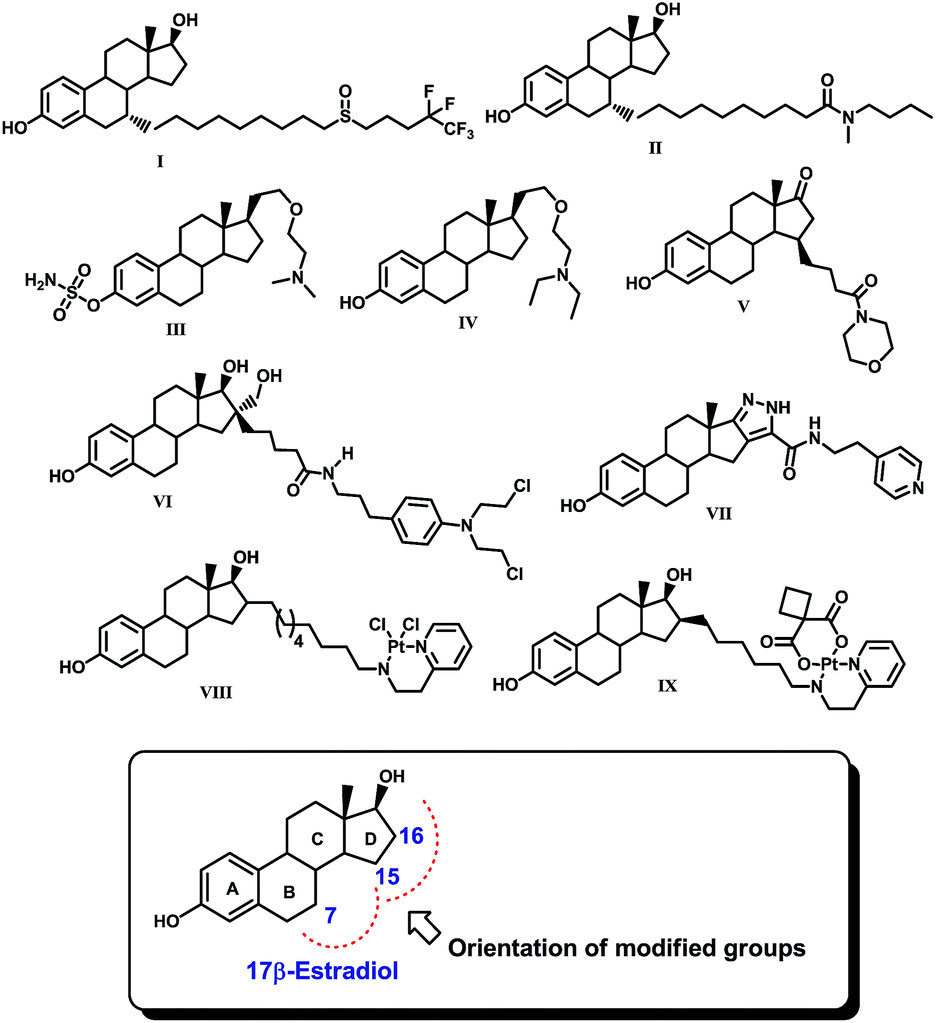 | ||
| Fig. 1 Some potential 17β-estradiol based anticancer drug molecules (I–IX) and 17β-estradiol. | ||
Since 17β-estradiol is a native ligand of the estrogen receptor (ER), a possible hypothesis for using estrane in these molecules could be targeted delivery of therapeutics selectively to the target site. It is interesting to note that all these compounds possess estradiol as the basic core, which has been modified by the incorporation of either an amino, amido or pentafluoroalkyl group or a platinum complex at position 7, 15, 16 or 17 of the steroid nucleus. These modifications were made in such a way that the modified groups could be accommodated spatially somewhere in the ring B and/or D region within the ligand binding pocket (LBP) of ER. However, in this approach, the incorporation of the estrane nucleus sometimes creates secondary complications due its intrinsic estrogen agonistic action. Therefore, a search for effective non-steroidal ligands for the estrogen receptor such as tamoxifen and other non-steroidal antiestrogens is warranted to address this issue.
Furthermore, a quinazoline nucleus, which is present in lapatinib (X), erlotinib (XI), gefitinib (XII) and vandetanib (XIII) applied for the treatment of cancer, has been well explored in the development of anticancer drugs Fig. 2.12–15 In our approach to devise potential non-steroidal anticancer agents especially for estrogen receptor positive breast cancer treatment, we have synthesized and evaluated 4-phenyl-5,6-dihydrobenzo[h]quinazoline derivatives for their anticancer potential. The designed molecules will have a quinazoline embedded tricyclic ring system analogous to rings A, B and C of estradiol and a phenyl ring bearing an alkylamino or ester group that is positioned to flank where the ring D of the estradiol core would have been, as presented in Fig. 1 and 2.
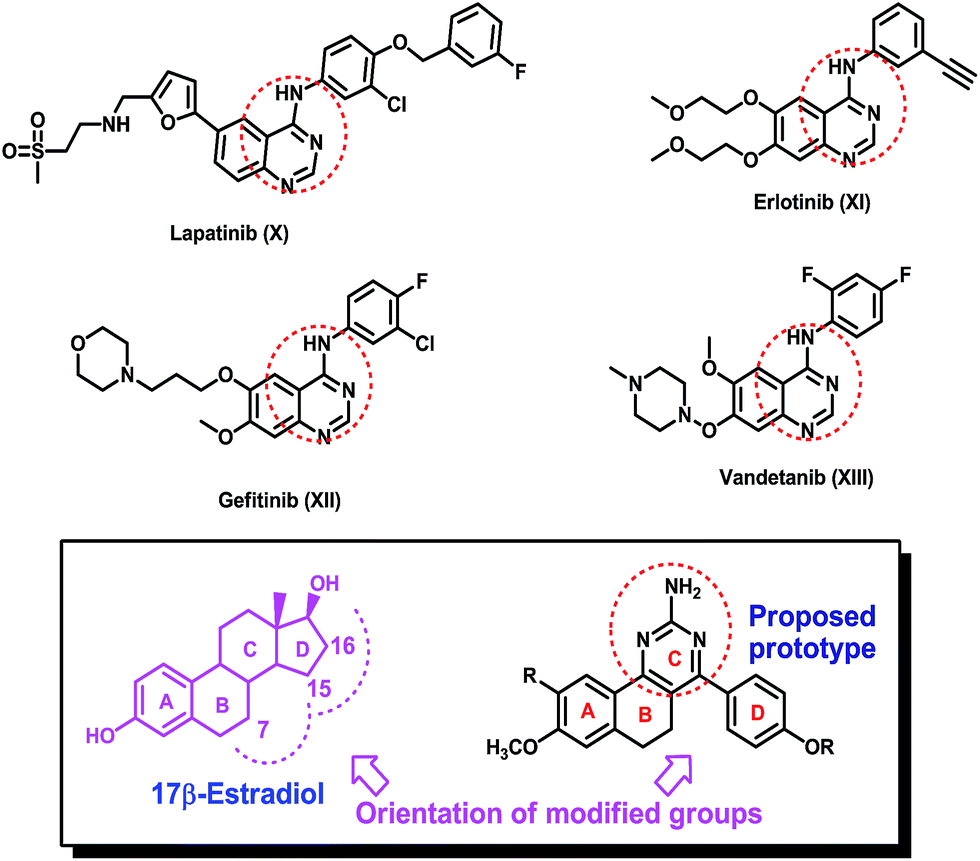 | ||
| Fig. 2 Quinazoline fragment-containing anticancer drugs, 17β-estradiol and the proposed prototype. | ||
Results and discussion
Chemistry
The synthesis of the designed prototype was started with the preparation of different chalcones (3), important intermediates for the target compounds. Compound 3 was initially synthesized from the reaction of 1-tetralones (1) and substituted benzaldehydes (2) using BF3–OEt2 in dioxane for 72 h at room temperature under dry reaction conditions as previously reported by us.16 However, attempts were made to find an alternate method for the preparation of 3 which could yield 3 in a short reaction time and would not require dry reaction conditions. For this purpose, we used N/10 HCl in an aqueous medium at room temperature which offered simple reaction conditions, a comparatively short reaction time (50 h vs. 72 h) and the formation of chalcone as the sole product. However, the yields of the product using this method were moderate. Further attempts were made to improve the yields and the best conversion was observed from reaction at 50 °C for 50 h, and these conditions were used for the chalcone synthesis (Scheme 1).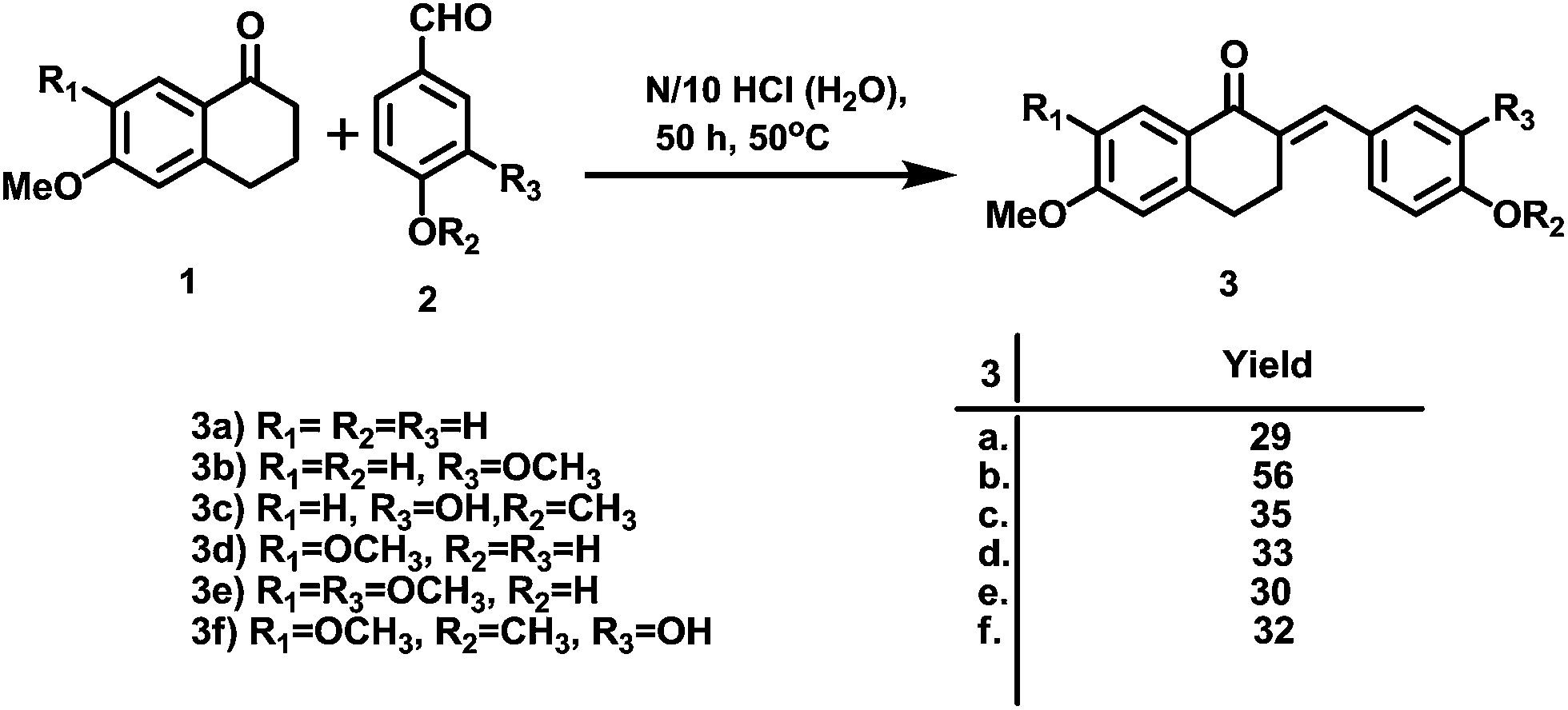 | ||
| Scheme 1 Synthesis of the chalcones (3). | ||
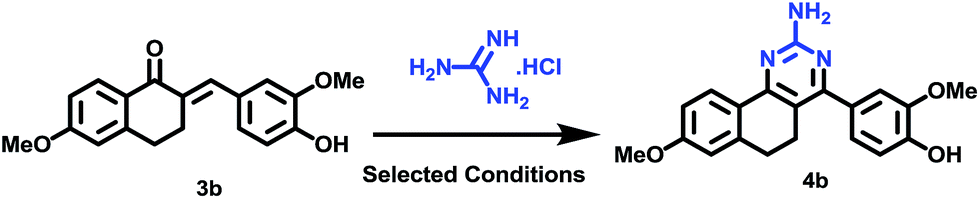
Subsequently, the targeted quinazoline analogs (4) were synthesized through the reaction of a chalcone (3) with guanidine hydrochloride. For this purpose, we explored the use of green solvents for synthesis of the target compounds in a short reaction time. In our attempts, we found choline chloride (ChCl) to be an appropriate green, non toxic, environmentally benign solvent for the synthesis of the highly substituted 5,6-dihydro-8-methoxybenzo[h]quinazolin-amine (4) Scheme 2. The use of choline chloride (ChCl) containing eutectic solvents has been reported for various reactions e.g. ChCl/urea supported conversion of aromatic boronic acids in phenol, ChCl/urea supported chemical fixation of carbon dioxide to provide cyclic carbonates, ChCl/glycine assisted biotransformations of ethyl valerate to butyl valerate and bromination of a substituted 1-aminoanthra-9,10-quinone, ChCl/L(−)-proline supported aldol reactions, a Knoevenagel condensation in ChCl/amino acid, Diels–Alder cycloadditions and Fischer indole annulations in ChCl/ZnCl2, and ring opening of epoxides in ChCl/SnCl2.17–24
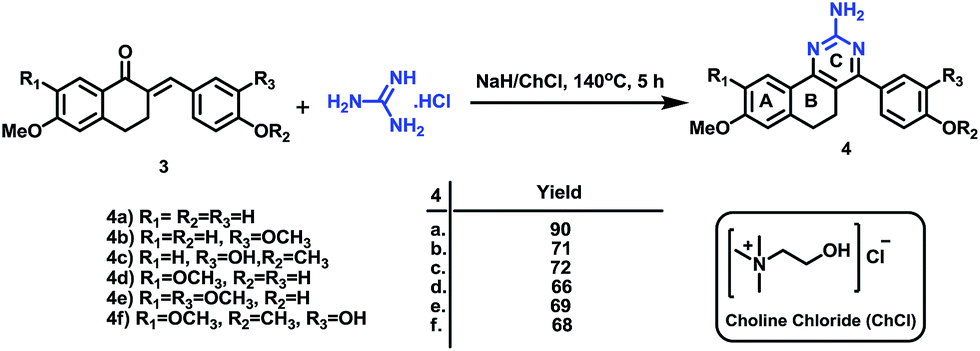 | ||
| Scheme 2 Synthesis of benzo[h]quinazolin-2-amines (4). | ||
Different reaction conditions were attempted using ChCl at different temperatures and molar ratios of reactants. The desired compounds were finally synthesized in good yield within 5 h by the use of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 molar ratio of reactants with NaH in ChCl at 140 °C (Table 1). Interestingly, as previously reported by us, the use of a conventional solvent such as dimethylformamide (DMF) under the reaction conditions requires a longer reaction time viz 36 h.16
:1.2 molar ratio of reactants with NaH in ChCl at 140 °C (Table 1). Interestingly, as previously reported by us, the use of a conventional solvent such as dimethylformamide (DMF) under the reaction conditions requires a longer reaction time viz 36 h.16
Furthermore, quinazoline 4a was modified to provide alkylamine substituted quinazolines (6) through the reaction of substituted alkylamine hydrochlorides (5) with 4 in the presence of anhydrous K2CO3 in chloroform–acetone (1:1).16 Similarly, alkylation of 4a was performed using bromoalkyl esters in the presence of potassium carbonate K2CO3 in dimethylformamide (DMF), which yielded long chain ester substituted quinazolines (8) in 90–94% yields (Scheme 3).16
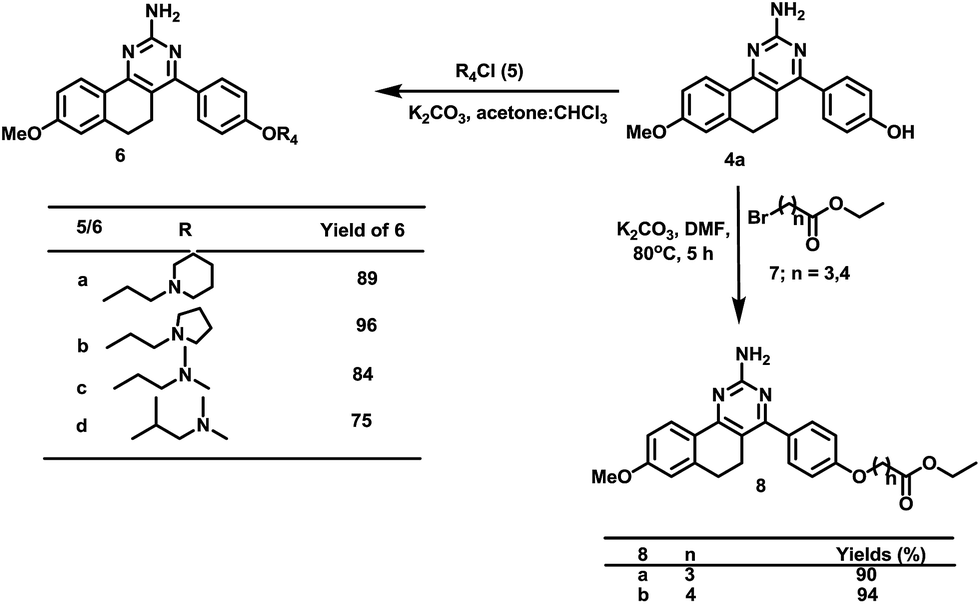 | ||
| Scheme 3 Synthesis of the benzo[h]quinazolin-2-amines derivatives (6 and 8). | ||
The synthesized compounds were characterized using different spectroscopic techniques viz NMR, IR, and mass spectrometry.
Biology
The synthesized compounds were investigated for their in vitro growth inhibition (anticancer) activity for cancer cells using a panel of cancer cell lines viz MCF-7 A549, FaDu and DU145 using a SRB assay. Some promising molecules (4a and 6d) were evaluated for their antitubulin activity using an in vitro tubulin polymerization assay. Furthermore, compound 6d was evaluated in a cell division cycle study using MCF-7 cells and for its capability to induce apoptosis in cancer cells (MCF-7 cells) using a PARP assay. In silico docking experiments for 4a and 6d with estrogen receptor-α and a tubulin protein were performed to verify the intended design of these molecules. The results of these studies are elaborated in the following text.In vitro growth inhibition of cancer cells
The compounds 4(a–f), 6(a–d), 8a and 8b were explored for their in vitro anticancer activity using various human carcinoma cell lines such as DLD1 (colorectal adenocarcinoma), A549 (lung carcinoma), FaDu (hypopharyngeal carcinoma), MCF-7 (ER + breast adenocarcinoma), and DU145 (prostate carcinoma) using a sulphorhodamin B assay. Tamoxifen was used as a positive control in this study. The anticancer activity of the tested compounds is described using the half maximal inhibitory concentration (IC50) value in Table 2.| Compounds | Carcinoma cell lines (IC50 in μM, mean ± SE) | ||||
|---|---|---|---|---|---|
| MCF-7 | DLD1 | A549 | DU145 | FaDu | |
| a Values are presented as the mean IC50 value ± SE of three independent experiments. The concentration of the compounds used for IC50 determination was obtained from 5 serial dilutions (2 fold) of the 20 μM starting concentration. The incubation period for the drug treated cells was 48 h.b Cell lines used: DLD1 (colorectal adenocarcinoma), A549 (lung carcinoma), FaDu (hypopharyngeal carcinoma), MCF-7 (ER + breast adenocarcinoma), DU145 (prostate carcinoma), [a] = not done. [b] although the compounds had been tested thrice in this cell line, value of one assay was >50, therefore it could not be included in calculation of mean and SE. | |||||
| 4a | 5.89 ± 1.22 | <1.50 | 1.86 ± 0.18 | <1.50 | [a] |
| 4b | >20 | <1.87 | >20 | [a] | [a] |
| 4c | >20 | 6.79 ± 1.14 | >20 | >20 | [a] |
| 4d | >20 | 2.96 | >20 | 5.25 | [a] |
| 4e | >20 | >20 | >20 | >20 | [a] |
| 4f | >20 | >20 | >20 | >20 | [a] |
| 6a | 7.09 ± 0.66 | 3.90 ± 0.82 | 7.97 ± 0.19 | 7.82 ± 1.17 | 7.59 ± 1.01 |
| 6b | 12.13 ± 0.54 | 6.64 ± 1.22 | 11.36 ± 0.54 | 10.41 ± 1.48 | 9.92 ± 0.94 |
| 6c | 7.84 ± 0.91 | 4.22 ± 0.94 | 8.05 ± 0.18 | 7.76 ± 1.41 | 7.51 ± 1.28 |
| 6d | 6.38 ± 0.37 | 3.63 ± 0.71 | 7.35 ± 0.11 | 6.96 ± 1.27 | 6.44 ± 0.55 |
| 8a | >20 | >20 | >20 | >20 | >20 |
| 8b | 8.03 ± 0.62 | 15.10 [b] | 12.99 ± 0.72 | >20 | >20 |
| Tamoxifen | 8.74 ± 1.17 | 14.65 | 10.37 ± 0.84 | 12.14 ± 0.62 | [a] |
Among hydroxyl derivatives 4a–f, compounds 4b and 4c which possess methoxy and hydroxy groups on ring D at positions 3 and 4 respectively presented anticancer activities in colon cancer cells at 1.87 and 6.79 μM concentrations, whereas 4e and 4f which have two methoxy groups on ring A along with methoxy and hydroxy groups on ring D were devoid of anticancer activity. Compound 4d which has two methoxy groups on ring A and a hydroxy group on ring D showed significant anticancer activity in colon as well as prostate cancer cells at IC50 values of 2.96 and 5.25 μM. Among others, compound 4a which had a methoxy group on ring A and a hydroxy group on ring D presented potent anticancer activity in MCF-7, DLD-1, A549 and DU145 cancer cells, at 5.89, 1.50, 1.86 and 1.50 μM concentrations respectively, compared to tamoxifen, a positive control.
In order to improve the biological activity, compound 4a was chosen for further modification. Interestingly, alkylation of 4a with different tertiary aminoalkyl groups usually present in antiestrogens such as tamoxifen, yielded compounds which showed significant anticancer activity invariably in all cancer cell lines within a range of IC50 values of 3.63–12.13 μM. Furthermore, compound 8b which has a five carbon long chain and an ester group showed anticancer activity in MCF-7, DLD1, and A549 cells at 8.03, 15.10 and 12.99 μM concentrations respectively, whereas another ester analog 8a was devoid of any activity up to a concentration of 20 μM.
Tubulin polymerization inhibition
Microtubule/tubulin dynamics interrupting molecules bind to the tubulin protein, which perturbs mitosis and arrests the growth of cells during interphase.25 Having growth inhibition activity data in our hands, we next wished to evaluate whether or not these molecules could inhibit the growth of cancer cells through inhibition of tubulin polymerization. For this purpose, compounds 4a and 6d were evaluated for their tubulin polymerization inhibition activity using the tubulin protein in an in vitro model. In this experiment, podophyllotoxin and nocodazole, standard tubulin polymerization inhibitors, were used as positive controls and DMSO as a negative control. Compounds 4a and 6d effectively inhibited tubulin polymerization at an IC50 value 2.92 and 5.97 μM respectively. Compound 6d presented antitubulin activity very close to that of podophyllotoxin (Table 3).| S. no. | Compound | IC50 (μM) |
|---|---|---|
| a Values are presented as the mean IC50 value ± SE of three independent experiments. | ||
| 1 | 4a | 5.97 ± 0.7 |
| 2 | 6d | 2.92 ± 0.16 |
| 3 | Podophyllotoxin (PDT) | 2.27 ± 0.6 |
| 4 | Nocodazole | 2.06 ± 0.07 |
Cell division cycle study of 6d
Furthermore, compound 6d was selected for a cell division cycle study using an estrogen receptor (ER) positive human breast cancer cell line (MCF-7). After 24 and 48 h incubation with 6d at 7 μM concentration (near the IC50 value), substantial accumulation of the cell population at the sub-G0 (apoptotic) and G0/G1 phases compared to the vehicle treated controls was observed in a time dependent manner (Fig. 3a and b). This was associated with a concomitant decrease in the cells at the S phase while there was no obvious change in the G2/M population.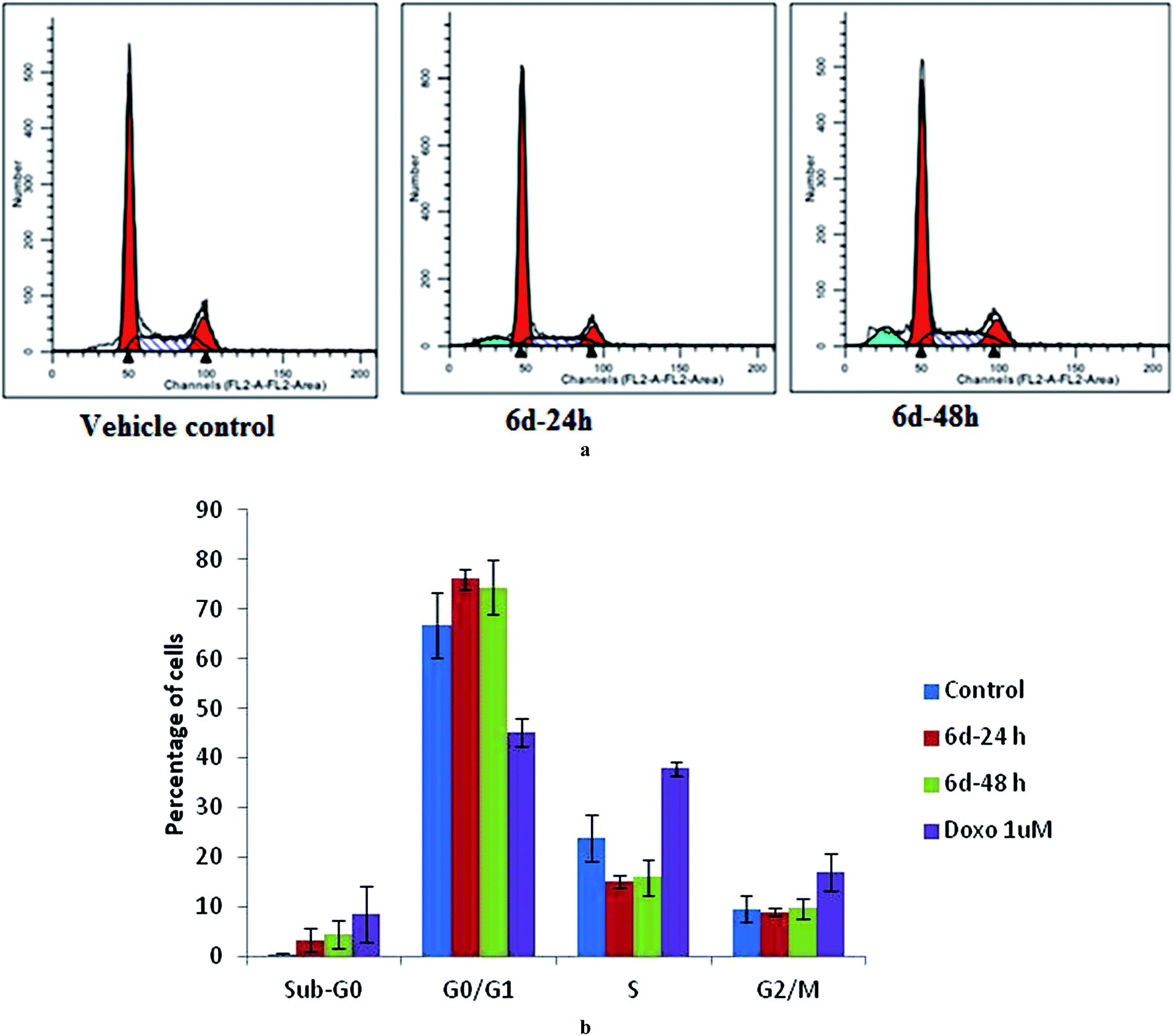 | ||
| Fig. 3 Effect of 6d on the cell division cycle (a) MCF-7 cells were treated with 6d at IC50 concentrations for 24 h and 48 h, stained with propidium iodide (PI), and were subjected to flow cytometry. (b) Histogram showing the average population of cells in various phases (G1, G2, S) of the cell cycle (mean ± S.E. of three independent assays, each performed in duplicate). #P < 0.05, *P < 0.001 compared with the vehicle treated controls. | ||
Cell apoptosis study
Following the anticancer, tubulin polymerization inhibition activity and cell cycle studies of 6d, we further investigated the ability of 6d to trigger apoptosis in MCF-7 cells using a western blot assay after probing with an anti-PARP antibody. Poly(ADP-ribose) polymerase (PARP) is a protein which is involved in a number of cellular processes including DNA repair and programmed cell death through the production of PAR, which stimulates mitochondria to release the apoptosis-inducing factor (AIF).26 In the present study, we monitored cleavage of poly(ADP-ribose) polymerase (PARP), which is considered as a marker of apoptosis. Consistent with the flow cytometry data, the western blot analysis of MCF-7 cell lysates revealed proteolysis of PARP in the 6d treated cells, confirming the induction of apoptosis (Fig. 4). In this experiment, doxorubicin (doxo), a potent anticancer drug was used as a positive control. | ||
| Fig. 4 6d induces apoptosis in MCF-7 cells. Lysates from the treated and untreated cells were subjected to immunoblotting after probing with an anti-PARP antibody. Marked cleavage of PARP was observed in the 6d treated cells in a time dependent manner. | ||
Docking study
To investigate the mechanism of action of the studied molecules, a molecular docking study was performed for 4a and 6d against the targets ER-α and tubulin using Autodock MGL tool v1.5.6. The docking study with estrogen receptor-α (ER-α) revealed that compounds 4a and 6d exhibit promising binding affinity within the tamoxifen binding site with a docking binding energy of −7.12 kcal mol−1 (Ki value 6.01 μM) and −5.55 kcal mol−1 (Ki value 84.85 μM) respectively (Fig. 5 and 6). The identified key residues for ligand–receptor interaction are ARG 394, THR 347 and GLY 420. The –NH2 group of the amino acid arginine interacts with the oxygen atom of the hydroxyl group of molecules 4a and 6d with a bond distance of 2.79 and 2.80 Å respectively. The compound 4a forms two additional H-bonds with the amino acid residues THR 347 and GLY 420, with bond distances of 2.07 and 1.7 Å. The results are summarized in Tables 4 and 5.| Compounds | Binding energy (kcal mol−1) | Ki value (inhibitory constant) | Binding pocket residues within a 4 Å region | Interacting residues and H-bond length (Å) | Number of rotatable bonds |
|---|---|---|---|---|---|
| Tamoxifen | −8.89 | 304.35 nM | MET343, LEU346, THR347, ALA350, ASP351, GLU353, TRP383, LEU384, LEU387, MET388, LEU391, ARG394, PHE404, GLU419, GLY420, MET421, ILE424, LEU428, GLY521, HIS524, LEU525 | ARG394: NH2–tamoxifen: O4 (2.8) & tamoxifen: O4–GLU353: OE2 (2.4) | 8 |
| 4a | −7.12 | 6.01 μM | MET343, LEU346, THR347, LEU349, ALA350, GLU353, LEU384, LEU387, ARG394, GLU419, GLY420, MET 421, ILE424GLY521, HIS 524, LEU525 | ARG394: NH2–4a: O (2.79572), 4a: H–THR347: OG1 (2.0738) & 4a: H–GLY420: O (1.73547) | 2 |
| 6d | −5.55 | 84.85 μM | MET343, LEU346, THR347, ALA350, ASP351, GLU353, TRP383, LEU384, LEU387, MET388, LEU391, ARG394, ILE424, GLY521, LEU525, MET528 | ARG394: NH2–6d: O (2.80547) | 6 |
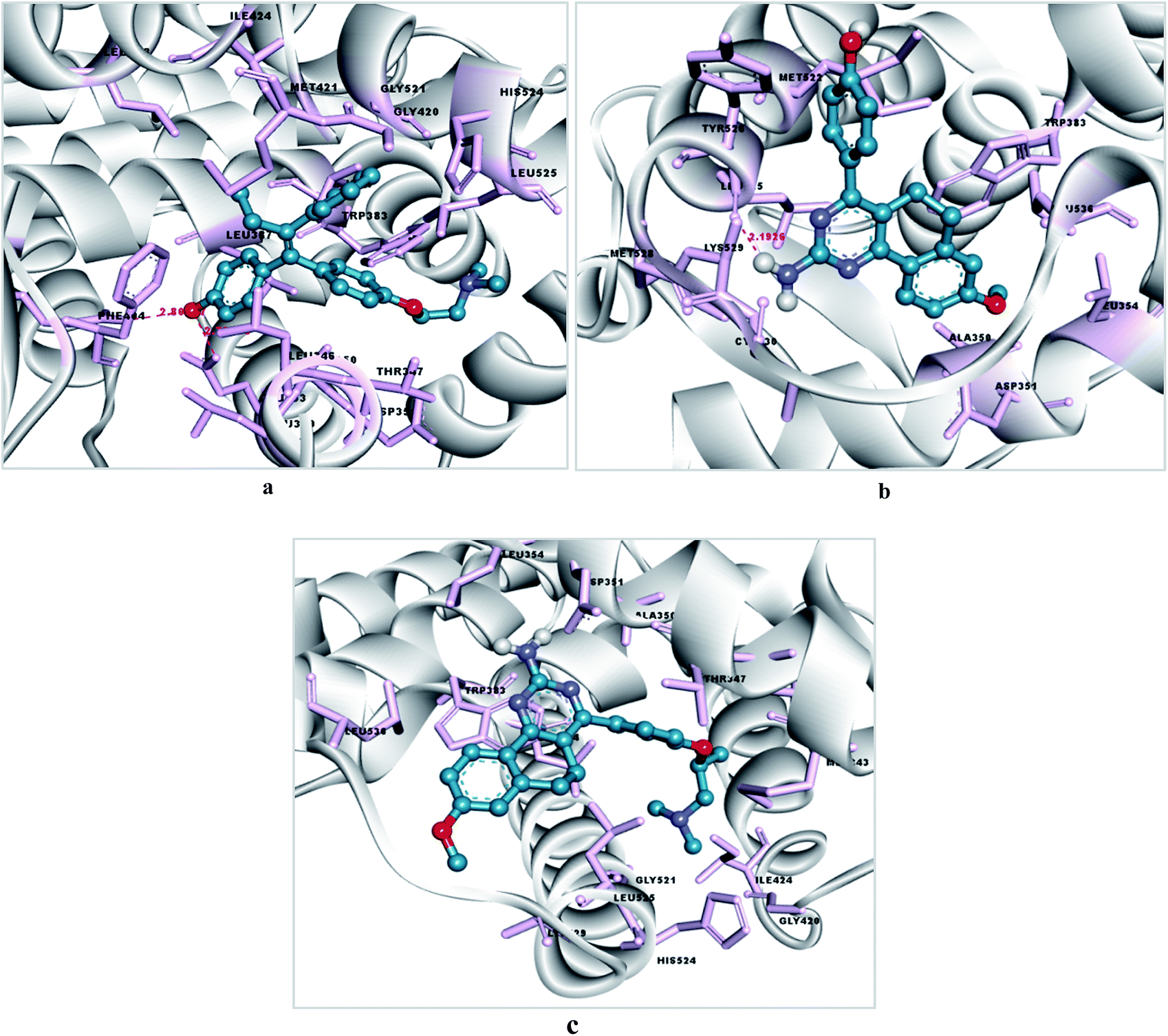 | ||
| Fig. 5 (a) Docking pose of tamoxifen within the LBP of estrogen receptor-α, (b) docking pose of 4a within the LBP of estrogen receptor-α (c) docking pose of 6d within the LBP of estrogen receptor-α. The residues are highlighted as pink sticks, the ligand is presented in a blue ball and stick form, and H-bonding is represented with pink dashed lines. | ||
 | ||
| Fig. 6 Binding affinities of tamoxifen, 4a and 6d with estrogen receptor-α (ERα). | ||
| Compounds | Binding energy (kcal mol−1) | Ki value (inhibitory constant) | Binding pocket residues within a 4 Å region | Interacting residues and H-bond length (Å) | Number of rotatable bonds |
|---|---|---|---|---|---|
| Podophyllotoxin | −5.16 | 165.71 μM | CYS241, LEU248, ALA250, ASP251, LYS254, LEU255, ASN258, VAL315, ALA316, LYS352, ALA354 | — | 5 |
| 4a | −5.59 | 79.52 μM | VAL238, THR239, CYS241, LEU242, LEU248, ALA250, LEU255, ASN258, ALA316, ALA317, ILE318, LYS352, THR353, ALA354 | 4a: H–ALA317: O (2.19842) & 4a: H–LYS352: O (2.22885) | 2 |
| 6d | −5.06 | 194.67 μM | VAL238, CYS241, LEU242, LEU248, ALA250, LYS254, LEU255, ASN258, MET259, THR314, ALA316, ILE347, PRO348, ASN349, ASN350, LYS352 | — | 6 |
Similarly, an interaction study of the molecules with the tubulin protein indicated that the control podophyllotoxin, 4a and 6d show good binding affinity within the colchicine binding site of the tubulin protein. 4a exhibits the highest docking binding energy of −5.59 kcal mol−1 (Ki value 79.52 μM) and forms two H-bonds with the amino acid residues ALA 317 and LYS 352, with bond distances of 2.198 and 2.229 Å. Whereas compound 6d showed a moderate docking binding energy of −5.06 kcal mol−1 (Ki value 194.67 μM) (Fig. 7 and 8).
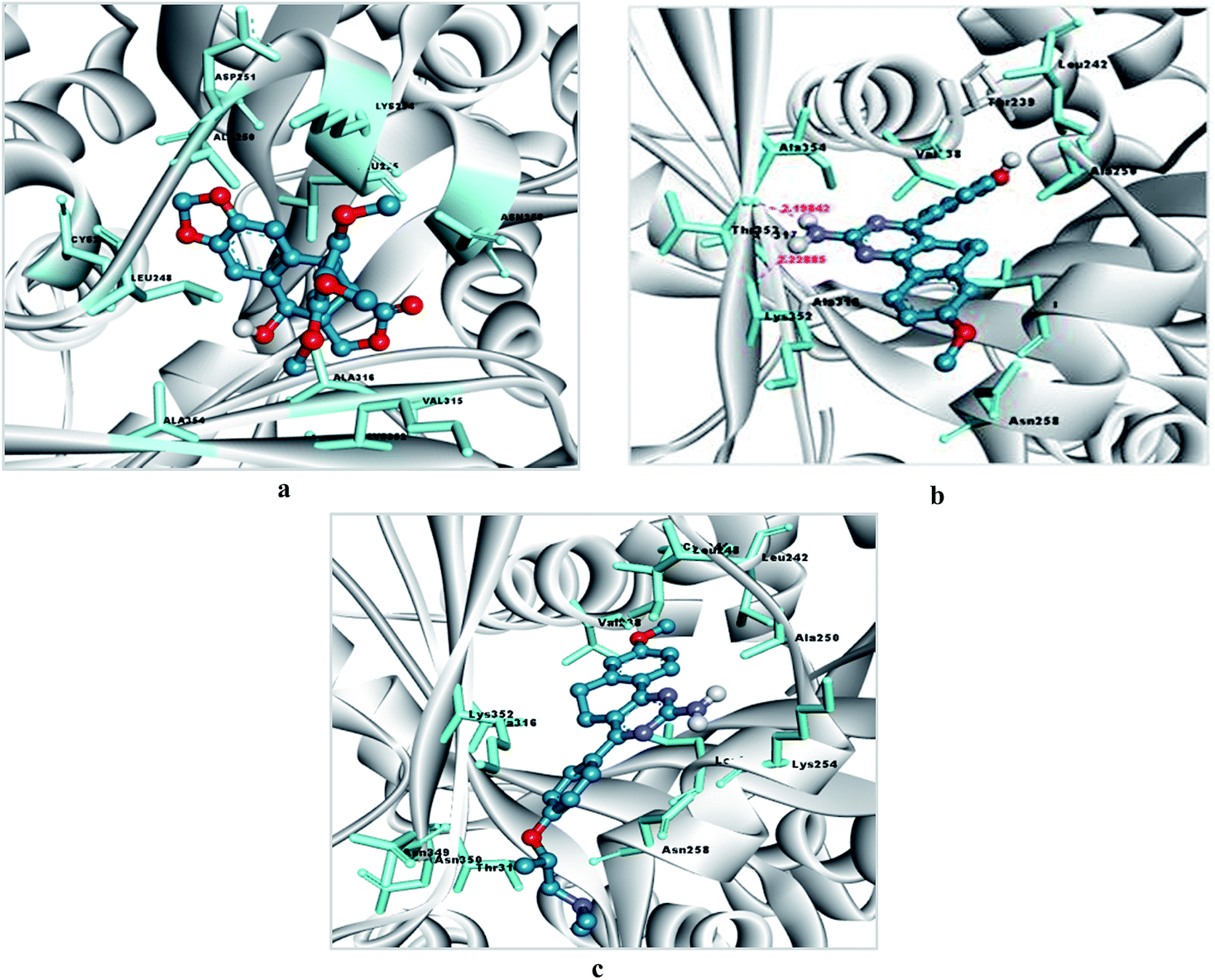 | ||
| Fig. 7 (a) Docking pose of podophyllotoxin within the binding site of tubulin, (b) docking pose of 4a within the binding site of tubulin, (c) docking pose of 6d within the binding site of tubulin. Binding pocket residues are highlighted with a cyan colored stick form, the ligand is presented in a blue ball and stick form, and H-bonding is represented with pink dashed lines. | ||
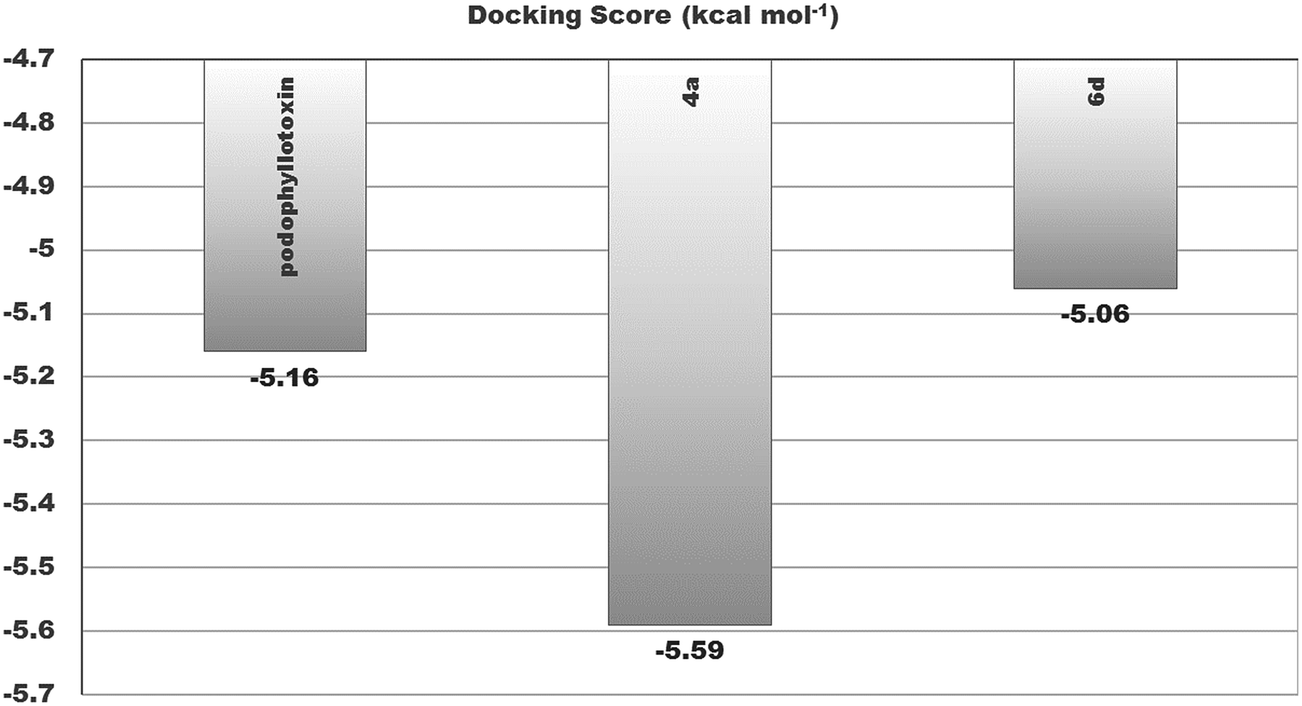 | ||
| Fig. 8 Binding affinities of tamoxifen, 4a and 6d with the binding site residues of β-tubulin. | ||
Conclusion
In conclusion, we have established a greener approach for the synthesis of substituted benzo[h]quinazoline analogs from chalcones and guanidine with good yields. The synthesized compounds presented significant anticancer activity using a panel of cancer cell lines. The cell apoptosis and tubulin polymerization inhibition activities of promising molecule 6d revealed that the anticancer activity of this compound is due to apoptosis of the cancer cells, which is very likely due to its tubulin polymerization inhibition activity. The in silico docking experiments of 4a and 6d showed that these molecules have the ability to interact with estrogen receptor-α and the colchicine binding site of the tubulin protein. The biological activity and in silico study results indicate that the structural arrangement of 6d causes it to interact with β-tubulin, resulting in its anticancer activity in general, and have estrogen receptor mediated activity in ER positive cancer cells (MCF-7 cells) in particular. Furthermore, structural modifications of these 4-phenyl-5,6-dihydrobenzo[h]quinazolines may lead to effective and ER selective anticancer lead molecules.Experimental section
General
The reagents and the solvents used in this study were of analytical grade and used without further purification. All the reactions were monitored using Merck aluminium thin layer chromatography (TLC, UV254 nm) plates. Column chromatography was carried out on silica gel (60–120 mesh). The melting points were determined using a Buchi melting point M560 apparatus in open capillaries and are uncorrected. Commercial reagents were used without purification. 1H and 13C NMR spectra were recorded on a Bruker WM-300 (300 MHz) using CDCl3 and DMSO-d6 as the solvents. Chemical shifts are reported in parts per million shift (δ-value) based on the middle peak of the solvent (CDCl3/DMSO-d6) as the internal standard. Signal patterns are described as s, singlet; bs, broad singlet; d, doublet; dd, double doublet; t, triplet; m, multiplet; brm, broad multiplet. Coupling constants (J) are given in Hertz. Infrared (IR) spectra were recorded on a Perkin-Elmer AX-1 spectrophotometer using KBr discs and the data are reported in wave numbers (cm−1). ESI mass spectra were recorded using a Shimadzu LC-MS after dissolving the compounds in acetonitrile and methanol.The synthesis of compounds 3a–f
Yellow solid, Rf: 0.3 (30% EtOAc in hexane); mp 172–173 °C; IR (KBr, νmax/cm−1): 3357 (NH2), 3489 (OH); 1H NMR (acetone-d6, 300 MHz, δ ppm): 2.92 (t, 2H, J = 6.0 Hz, CH2), 3.11 (t, 2H, J = 6.0 Hz, CH2), 3.88 (s, 3H, OCH3), 6.90 (m, 4H, ArH), 7.39 (d, 2H, J = 8.4 Hz, ArH), 7.68 (s, 1H, CH), 7.97 (d, 1H, J = 8.4 Hz, ArH), 9.35 (s, 1H, OH); 13C NMR (acetone-d6, 75 MHz, δ ppm): 27.60, 29.30, 55.58, 112.66, 113.87, 116.05 (2 × C), 127.50, 127.64, 130.49, 132.36 (2 × C), 133.54, 136.02, 146.27, 159.04, 164.03, 185.96; ESI-MS (C18H16O3): m/z: 281 [M + H]+.
The synthesis of compounds 4a–f
Off-white solid, Rf: 0.17 (10% methanol in chloroform); mp 263–265 °C; IR (KBr, νmax/cm−1): 3357 (NH2), 3489 (OH); 1H NMR (DMSO-d6, 300 MHz, δ ppm): 2.74 (bs, 2 × CH2), 3.80 (s, 3H, OCH3), 5.74 (s, 1H, OH), 6.31 (s, 2H, NH2), 6.83–6.92 (m, 4H, ArH), 7.42 (d, 2H, J = 8.4 Hz, ArH), 8.09 (d, 1H, J = 8.4 Hz, ArH); 13C NMR (DMSO-d6, 75 MHz, δ ppm): 24.71, 28.94, 56.09, 113.38, 113.50, 113.77, 115.52 (2 × C), 126.86, 127.63, 129.97, 131.14 (2 × C), 142.19, 158.86, 160.50, 161.78, 162.81, 165.07; ESI-MS (C19H17N3O2): m/z = 320.4 [M + H]+.
The synthesis of compounds 6a–d
Off-white solid; Rf: 0.23 (10% methanol in chloroform); mp 152–155 °C; IR (KBr, νmax/cm−1): 3396 (NH2); 1H NMR (CDCl3, 300 MHz, δ ppm): 1.26 (bs, 2H, CH2), 1.46 (bs, 4H, 2 × CH2), 2.54 (bs, 4H, 2 × NCH2), 2.77–2.86 (bs, 6H, NCH2 & 2 × CH2), 3.85 (s, 3H, OCH3), 4.17 (t, 2H, OCH2), 5.00 (s, 2H, NH2), 6.73 (d, 1H, J = 2.1 Hz, ArH), 6.86 (d, 1H, J = 2.4 Hz, ArH), 6.89 (d, 2H, J = 2.1 Hz, ArH), 7.50 (d, 2H, J = 8.7 Hz, ArH), 8.20 (d, 1H, J = 8.7 Hz, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 24.52, 24.73, 26.24 (2 × C), 29.16, 55.43 (2 × C), 55.72, 58.20, 66.41, 112.95, 113.24, 114.72 (2 × C), 115.43, 126.67, 127.83, 130.54 (2 × C), 131.41, 141.92, 159.79, 161.36, 161.94, 161.98, 165.04; ESI-MS (C26H30N4O2): m/z = 431.5 [M + H]+.
The synthesis of compounds 8a and 8b
Light yellow solid; Rf: 0.26 (30% acetone in hexane); mp 148–149 °C; IR (KBr, νmax/cm−1): 3474 (NH2), 1728 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (DMSO-d6, 300 MHz, δ ppm): 1.67 (t, 3H, J = 6.9 Hz, CH3), 1.97 (t, 2H, J = 6.9 Hz, CH2), 2.45 (p, 2H, J = 7.2 Hz, CH2), 2.72 (bs, 4H, 2 × CH2), 3.78 (s, 3H, OCH3), 4.00 (m, 4H, 2 × OCH2), 6.31 (bs, 2H, NH2), 6.88 (d, 1H, J = 2.1 Hz, ArH), 6.90 (d, 1H, J = 8.7 Hz, ArH), 6.98 (d, 2H, J = 8.7 Hz, ArH), 7.49 (d, 2H, J = 8.7 Hz, ArH), 8.08 (d, 1H, J = 8.7 Hz, ArH); 13C NMR (DMSO-d6, 75 MHz, δ ppm): 14.92, 24.61, 25.05, 28.86, 31.03, 56.08, 60.79, 67.46, 113.41, 113.52, 113.94, 114.67 (2 × C) 126.72, 127.68, 131.07 (2 × C), 131.53, 142.22, 159.71, 160.64, 161.85, 162.79, 164.75, 173.48; ESI-MS (C25H27N3O4): m/z = 434.2 [M + H]+.
O); 1H NMR (DMSO-d6, 300 MHz, δ ppm): 1.67 (t, 3H, J = 6.9 Hz, CH3), 1.97 (t, 2H, J = 6.9 Hz, CH2), 2.45 (p, 2H, J = 7.2 Hz, CH2), 2.72 (bs, 4H, 2 × CH2), 3.78 (s, 3H, OCH3), 4.00 (m, 4H, 2 × OCH2), 6.31 (bs, 2H, NH2), 6.88 (d, 1H, J = 2.1 Hz, ArH), 6.90 (d, 1H, J = 8.7 Hz, ArH), 6.98 (d, 2H, J = 8.7 Hz, ArH), 7.49 (d, 2H, J = 8.7 Hz, ArH), 8.08 (d, 1H, J = 8.7 Hz, ArH); 13C NMR (DMSO-d6, 75 MHz, δ ppm): 14.92, 24.61, 25.05, 28.86, 31.03, 56.08, 60.79, 67.46, 113.41, 113.52, 113.94, 114.67 (2 × C) 126.72, 127.68, 131.07 (2 × C), 131.53, 142.22, 159.71, 160.64, 161.85, 162.79, 164.75, 173.48; ESI-MS (C25H27N3O4): m/z = 434.2 [M + H]+.
In vitro cancer cell growth inhibition assay
The in vitro anticancer activity of the synthesized compounds was studied using a sulphorhodamine B (SRB) dye based plate assay. In brief, 104 cells per well were added into 96-well culture plates and incubated at 37 °C with a 5% carbon dioxide concentration. After overnight incubation of the cells, solutions from serial dilution of the synthesized compound were added to the wells. Untreated cells served as a control. After 48 h, the cells were fixed with ice-cold tri-chloroacetic acid (50% w/v, 100 mL per well), stained with SRB (0.4% w/v in 1% acetic acid, 50 mL per well), washed and air-dried. The bound dye was solubilized with 10 mM tris base (150 mL per well) and the absorbance was read at 540 nm on a plate reader. The cytotoxic effect of the compound was calculated as % inhibition in cell growth as per the formula: [1 − (absorbance of drug treated cells/absorbance of untreated cells) × 100]. Determination of the IC50 (50% inhibitory concentration) was based on the dose–response curves.Tubulin polymerization assay
To a pre-warmed 96 well plate, the compounds were added at a conc. of 10, 2, 0.4 and 0.08 μg mL−1. Paclitaxel was used as a standard at a conc. of 10 μM. The tubulin protein provided in the tubulin polymerization kit from Cytoskeleton, Inc. was added to a pre-prepared ice cold tubulin polymerization buffer [80 mM PIPES pH 6.9, 2 mM MgCl2, 0.5 mM EGTA] immediately before use and was kept on ice. The ratio at which the tubulin protein was mixed with the tubulin polymerization buffer is 200 μL of protein with 420 μL of tubulin polymerization buffer, which gave a final concentration of 3 mg mL−1 tubulin in 80 mM PIPES pH 6.9, 2 mM MgCl2, 0.5 mM EGTA.50 μL of this diluted tubulin solution was added to each well containing the tested compound at different concentrations and immediately the plate was read at 37 °C with pre-set kinetic parameters at a 340 nm wavelength for a period of 60 min.
The increase in absorbance at the pre-set kinetic parameters (340 nm, 37 °C over 60 min period of time) was graphically obtained as a polymerization curve and the Vmax values obtained using Skanit software 4.0 were used to interpret the results for analyzing the effect of the compounds on tubulin polymerization.
Cell cycle analysis
Cell cycle distribution was measured in a concentration and time dependent manner using flow cytometric analysis of PI-stained cellular DNA, as described earlier. Briefly, MCF-7 cells (8 × 105 per well) were seeded in 60 mm tissue culture dishes and grown overnight (37 °C, 5% CO2). The compound treated cells were harvested using trypsinization and fixed (30 min, 4 °C) with ice-cold 70% ethanol at the indicated time points. The pellets were washed with PBS and re-suspended in a solution containing PI (20 μg mL−1), Triton X-100 (0.1%) and RNase (0.1 mg mL−1) in PBS. After incubation (45 min, in the dark, 37 °C), the cells were analysed using a FACS Calibur flow cytometer (BD Biosciences). The distribution of cells in different phases of the cell cycle was calculated using “Cell Quest” software.Western blot assay
Cells were grown overnight in 60 mm tissue culture dishes and were exposed to the vehicle and test molecule for 24 and 48 h at the IC50 concentration. The cells were then harvested and lysed with a M-PER reagent (Thermo Scientific) supplemented with protease and phosphatase inhibitors. After centrifugation at 12000 rpm, the supernatant was collected and the protein quantity was measured using a BCA protein assay kit (Thermo Scientific). Equal amounts of proteins (20 μg) were resolved using 8% SDS-PAGE, transferred onto a PVDF membrane and probed overnight with anti-PARP (Cell Signaling Technology) and anti-actin (Sigma-Aldrich) antibodies. After incubation with a HRP-conjugated secondary antibody for 1 h at room temperature, ECL solution (Bio-Rad) was added to the membrane and the luminescence was detected using a Chemidoc XRS+ system (Bio-Rad).
Ligand and protein preparation for the docking experiments
The molecular docking of control inhibitors tamoxifen and colchicine, and compounds 4a–f, 6a–d, 8a and 8b was executed using the software Autodock v 1.5.6.23 The 3.5 Å 3D crystallographic structures of estrogen receptor alpha & tubulin were retrieved through the Brookhaven Protein DataBank (PDB) (http://www.pdb.org) (PDB ID:3ERT and 4O2B). The ligand and receptor preparations were done using Discovery studio v3.5 (Accelrys, Inc., San Diego, CA, USA). The receptor grid was generated using the ligands tamoxifen and colchicine co-crystallized in the receptor. The standardization of the software was done by performing a redocking study of the bound inhibitor. Visualization of the docked conformation was performed using pymol.
Acknowledgements
The authors thank the Director of CSIR-CIMAP and CSIR-CDRI for financial support. The work was carried out under project ChemBio BSC-203, associated with AG, and GAP 0115 (sponsored by DST, India) associated with JS. AS and FK acknowledge the Science & Engineering Research Board (SERB), Department of Science & Technology (DST), New Delhi, India for financial support through the GAP-260 project (Sanction No. SR/FT/LS-25/2010; 02/05/2012).Notes and references
- http//:www.healthindia.com “Diseases & Conditions World Cancer Day 2014: New cancer cases to increase five-fold by 2025”.
- R. Siegel, J. Ma, Z. Jou and A. Jemal, Cancer statistics 2014, Ca-Cancer J. Clin., 2014, 64, 9 CrossRef PubMed.
- http://www.who.int/mediacentre/factsheets/fs297/en/:Fact%20sheet%20N°297.
- K. l. Dao and R. N. Hanson, Bioconjugate Chem., 2012, 23, 2139 CrossRef CAS PubMed.
- J. D. Croxtall and K. McKeage, Drugs, 2011, 71, 363 CrossRef CAS PubMed.
- A. E. Wakeling, M. Dukes and J. Bowler, Cancer Res., 1991, 51, 3867 CAS.
- A. Gupta, B. S. Kumar and A. S. Negi, J. Steroid Biochem., 2013, 13, 7242 Search PubMed.
- (a) J. Messinger, H. H. Thole, B. Husen, M. Weske, P. Koskimies and L. Pirkkala, U.S. Pat. Appl. Publ, US 20060281710 A1 20061214, 2006; (b) J. Messinger, H. H. Thole, B. Husen, M. Weske, P. Koskimies, L. Pirkkala and M. Weske, PCT Int. Appl, WO 2006125800 A1 20061130, 2006.
- (a) C. V. Themsche, S. Parent, V. Leblanc, C. Descoteaux, A. M. Simard, G. Berube and E. Asselin, Endocr.–Relat. Cancer, 2009, 16, 1185 CrossRef PubMed; (b) K. Brasseur, V. Leblanc, F. Fabi, S. Parent, C. Descoteaux, G. Berube and E. Asselin, Endocr. J., 2013, 154, 2281 CrossRef CAS PubMed.
- G. M. Allan, C. Bubert, N. Vicker, A. Smith, H. J. Tutill, A. Purohit, M. J. Reed and B. V. L. Potter, Mol. Cell. Endocrinol., 2006, 248, 204 CrossRef CAS PubMed.
- A. Gupta, P. Saha, C. Descoteaux, V. Leblanc, E. Asselin and G. Berube, Bioorg. Med. Chem. Lett., 2010, 20, 1614 CrossRef CAS PubMed.
- C. E. Geyer, J. Forster, D. Lindquist, S. Chan, C. G. Romieu, T. Pienkowski, A. Jagiello-Gruszfeld, J. Crown, A. Chan, B. Kaufman, D. Skarlos, M. Campone, N. Davidson, M. Berger, C. Oliva, S. D. Rubin, S. Stein and D. Cameron, N. Engl. J. Med., 2006, 355, 2733 CrossRef CAS PubMed.
- E. Petit-Jean, T. Buclin, M. Guidi, E. Quoix, B. Gourieux, L. A. Decosterd, A.-C. Gairard-Dory, G. Ubeaud-Séquier and N. Widmer, Ther. Drug Monit., 2015, 37, 2 CrossRef CAS PubMed.
- T. Araki, H. Yashima, K. Shimizu, T. Aomori, T. Hashita, K. Kaira, T. Nakamura and K. Yamamoto, Clin. Med. Insights: Oncol., 2012, 6407 Search PubMed.
- (a) S. Karras, P. Anagnostis and G. E. Krassas, Expert Opin. Drug Metab. Toxicol., 2014, 10, 469 CrossRef CAS PubMed; (b) A. D. Luca, A. D’Alessio, M. R. Maiello, M. Gallo, S. Bevilacqua, D. Frezzetti, A. Morabito, F. Perrone and N. Normanno, Expert Opin. Invest. Drugs, 2014, 23, 1295 CrossRef PubMed.
- H. K. Maurya, R. Verma, S. Alam, S. Pandey, V. Pathak, S. Sharma, K. K. Srivastava, A. S. Negi and A. Gupta, Bioorg. Med. Chem. Lett., 2013, 23, 5844 CrossRef CAS PubMed.
- http://www.en.wikipedia.org/wiki/Choline.
- C. Rub and B. Konig, Green Chem., 2012, 14, 2969 RSC.
- A. Zhu, T. Jiang, B. Han, J. Zhang, Y. Xie and X. Ma, Green Chem., 2007, 9, 169 RSC.
- S. B. Phadtare and G. S. Shankarling, Green Chem., 2010, 12, 458 RSC.
- P. Moriel, E. J. García-Suárez, M. Martínez, A. B. García, M. A. Montes-Morán, V. Calvino-Casilda and M. A. Bañares, Tetrahedron Lett., 2010, 51, 4877 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Green Chem., 2002, 4, 24 RSC.
- R. C. Morales, V. Tambyrajah, P. R. Jenkins, D. L. Davies and A. P. Abbott, Chem. Commun., 2004, 158 RSC.
- N. Azizi and E. Batebi, Catal. Sci. Technol., 2012, 2, 2445 CAS.
- A. S. Negi, Y. Gautum, S. Alam, D. Chanda, S. Luqman, J. Sarkar, F. Khan and R. Konwar, Bioorg. Med. Chem., 2015, 23, 373 CrossRef CAS PubMed.
- S. W. Yu, S. A. Andrabi, H. Wang, N. S. Kim, G. G. Poirier, T. M. Dawson and V. L. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18314 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra24429c |
| ‡ CIMAP communication No. CIMAP/PUB/2015/02. |
| This journal is © The Royal Society of Chemistry 2016 |