
Step-by-step deposition of type B gelatin and tannic acid displays a peculiar ionic strength dependence at pH 5†
Abstract
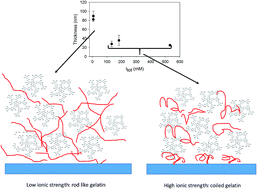
Tannic acid (TA), among other polyphenols, interacts strongly with proteins, in particular proline rich proteins, a mechanism which is at the origin of mouth astringency. Among such proline rich proteins are salivary rich proteins and collagen or gelatin. The formation of protein–polyphenol complexes is rarely, with some exceptions, of utility in materials science. However, when the complexation is controlled on surface templates, using the layer-by-layer deposition method, useful materials such as membranes for controlled separations or controlled polyphenol release can be obtained. The performance of such protein–TA containing films can only be improved through a better understanding of the parameters controlling the film deposition. Indeed it is widely believed and partially demonstrated that the protein–polyphenol interactions are driven through an interplay of hydrophobic interactions and hydrogen bonding. In this article, the deposition of (gelatin–TA)n films is investigated as a function of the ionic strength using a combination of characterization techniques: quartz crystal microbalance with dissipation monitoring (QCM-D), atomic force microscopy (AFM), Fourier transform spectroscopy in the attenuated total reflection mode (FTIR-ATR) and cyclic voltammetry (CV). It is shown that the film thickness and the amount of deposited film decreases strongly when the ionic strength is increased below about 50–100 mM but these film properties become almost ionic strength independent above 100 mM. The transition dynamics between these two growth regimes is investigated by putting the as prepared films in contact with a solution of different ionic strength.
 Please wait while we load your content...
Please wait while we load your content...

