
Solvate ionic liquid electrolyte with 1,1,2,2-tetrafluoroethyl 2,2,2-trifluoroethyl ether as a support solvent for advanced lithium–sulfur batteries†
Abstract
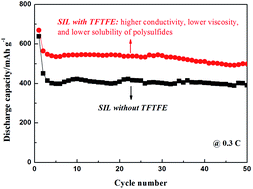
1,1,2,2-Tetrafluoroethyl 2,2,2-trifluoroethyl ether (TFTFE) was added to a solvate ionic liquid (SIL) based on glyme-lithium salt as a support solvent. The fluorinated ether improves cycle and rate capability of Li–S cells. The key role that TFTFE played in the cell system with SIL is an ionic conduction-enhancing ingredient, especially for high-rate cycle environment.
 Please wait while we load your content...
Please wait while we load your content...

