
Electrogenerated chemiluminescence of the tris(2,2′-bipyridine)ruthenium(ii)/aliphatic amine system: a universal effect of perchlorate salts†
Abstract
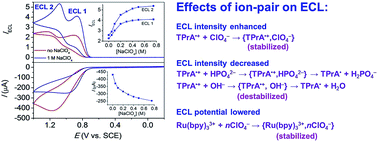
Sensitivity is a key parameter for electrogenerated chemiluminescence (ECL) analysis. We describe here a simple way to enhance the ECL of Ru(bpy)32+ (bpy = 2,2′-bipyridine) by adding concentrated perchlorate salts. Observations are made for a variety of aliphatic amines as coreactants in a wide pH range from pH 5 to 12 with ECL enhanced by 1.5–6.6 times for different ECL routes. By contrast, the addition of other salts such as nitrate, sulfate and chloride decreases ECL to some extent. Experimental studies suggest that perchlorate anions could stabilize the coreactant cation radicals by formation of ion pairs, which allows ECL to occur in a thicker reaction layer by a longer travel distance of these intermediates. However, other anions may get involved in detrimental redox processes. For example, nitrate and sulfate may directly react with the highly reducing coreactant free radicals or Ru(bpy)3+ intermediates, and chloride may be concomitantly oxidized at the electrode surface during ECL emission generating highly reactive HClO/ClO− intermediates. This study is important for both mechanistic fundamentals and the realistic applications of ECL.
 Please wait while we load your content...
Please wait while we load your content...

